
肾茶饮片的质量标准研究
2022-08-15 03:57林青华张永怡
中国民族民间医药 2022年14期
林青华 张永怡 周 慧 蒋 林*
1.广西中医药大学药学院,广西 南宁 530200;2.广西中医药大学广西壮瑶药工程技术研究中心,广西 南宁 530200
肾茶,又名猫须草,为唇形科植物肾茶Clerodendranthusspicatus(Thunb)C.Y.Wu ex H.W.Li的干燥地上部分,具有清热祛湿、排石通淋的功效[1]。肾茶含有黄酮、酚类、萜类、挥发油等多种化学成分,具有调节肾功能、利尿排石、抗炎、抗菌等多种药理活性[2-3],开发前景广阔。然而,肾茶饮片的质量标准并不完善,其中安徽[4]、云南[5]、四川[6]的饮片标准缺失“灰分”和“含量测定”,湖南[7]的饮片标准缺失“水分”“灰分”和“含量测定”,福建[8]、宁夏[9]的饮片标准仅有“性状”要求,这不利于质量控制和产品开发。本文按2020年版《中国药典》(四部)通则(下文简称药典)中的相关规定[10],对不同产地的10批肾茶饮片进行定性鉴别与定量研究,以期为完善肾茶饮片的质量标准提供参考与依据。
1 材料
1.1 药材 10批肾茶药材经广西中医药大学药用植物教研室朱意麟老师鉴定为唇形科植物肾茶Clerodendranthusspicatus(Thunb)C. Y.Wu ex H. W. Li的干燥地上部分,具体见表1。迷迭香酸对照品(批号MUST-19053107,纯度>98.0%)购于中国科学院成都生物研究所。
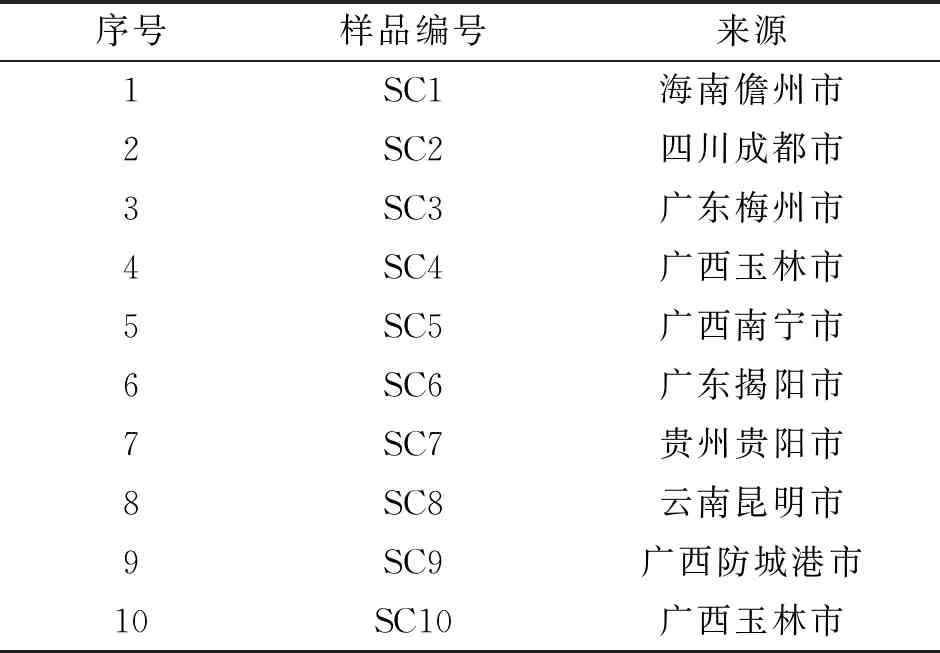
表1 10批肾茶药材编号与来源
1.2 仪器与试剂 Agilent-1260高效液相色谱仪(美国Agilent公司),Ultimate®XB-C18色谱柱[月旭科技(上海)股份有限公司],乙腈[赛默飞世尔科技(中国)有限公司],Direct-Q5UV超纯水仪一体机(广西南宁博美生物科技有限公司),DMB-1223型生物显微镜(麦克奥迪实业集团有限公司),ME204E型电子分析天平(梅特勒-托利多仪器有限公司),KQ-500E超声波清洗仪(昆山市超声仪器有限公司),硅胶G板(青岛海洋化工有限公司),甲醇、甲苯、甲酸、乙酸乙酯、水合氯醛、磷酸等均为分析纯。
2 方法与结果
2.1 性状鉴别 10批肾茶药材喷淋适量清水软化,切长段,干燥;按药典“0212药材和饮片检定方法”对其进行描述。本品为不规则的段、茎、叶混合。茎呈四棱形或类圆形,直径0.2~1.5 cm,外表皮紫褐色,有细纵线,有的有分支,切面周围黄白色至浅绿色,髓部类白色。叶对生,褐绿色,多破碎,完整者展平后呈卵形或卵状披针形,长2~5 cm,宽1~3 cm,两面呈黄绿色或暗绿色,叶脉紫褐色,均有小柔毛,先端尖,基部楔形,中部以上叶片边缘锯齿状,叶柄长约2 cm。轮伞花序偶见。气微,味微苦。具体如图1所示。

图1 肾茶饮片图
2.2 显微鉴别 按药典“2001显微鉴别法”对肾茶饮片的粉末进行鉴别。本品粉末呈绿褐色。表皮细胞类圆形或类方形,可见不定式气孔。非腺毛较多,单细胞,散在,多碎断,完整者基部细胞粗短膨大,壁较厚,有疣状突起,长85~175 μm。薄壁细胞内含草酸钙簇晶,常排列成行,也有的单个散在,直径8~18 μm。可见螺纹或环纹导管,以螺纹导管为主,常见于叶肉细胞中。偶见棕色块,类圆形,颜色深浅不一。如图2所示。

1.表皮细胞;2.草酸钙蔟晶;3.纤维组织;4、5.导管;6.气孔;7.非腺毛;8.腺毛
2.3 薄层鉴别 取肾茶饮片粗粉0.5 g,加甲醇10 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液。取迷迭香酸对照品,加甲醇制成每1 mL含0.2 mg的溶液,作为对照品溶液。照药典“0502薄层色谱法”规定,吸取对照品溶液、供试品溶液各5 μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲酸(5∶3∶0.8)为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视。结果发现,各供试品、对照品在硅胶G板的相应位置上,显相同颜色的荧光斑点,但各样品迷迭香酸的斑点深浅不一,说明其含量不尽相同。如图3所示。

图3 肾茶饮片供试品(1~10)与迷迭香酸对照品(R)在365 nm处的薄层色谱图
2.4 水分、灰分和浸出物的测定 按照药典“0832水分测定法(烘干法)”“2302灰分测定法”“2201浸出物测定法(水溶性浸出物,热浸法)”对其水分、总灰分、酸不溶性灰分及水溶性浸出物进行测定。检测结果,具体见表2。根据极值上、下浮动20%的原则,拟定肾茶饮片的水分、总灰分和酸不溶性灰分含量分别不得超过15.0%、11.0%、0.6%,水溶性浸出物含量不得低于17.0%。

表2 肾茶饮片水分、总灰分、酸不溶性灰分与水溶性浸出物的含量
2.5 含量测定
2.5.1 对照品溶液的制备 取迷迭香酸适量,精密称定,加50%甲醇制成每1 mL含0.1 mg的溶液,即得。
2.5.2 供试品溶液的制备 取肾茶饮片粉末(过五号筛)约0.25 g,精密称定,置具塞锥形瓶中,精密加50%甲醇25 mL,密塞,称定重量,超声处理0.5 h,放冷,再称定重量,用50%甲醇补足损失重量,摇匀,滤过,取续滤液,即得。
2.5.3 色谱条件 Ultimate®XB-C18色谱柱(4.6 mm×250 mm,5 μm),乙腈-0.1%磷酸水溶液(25∶75)为流动相,流速1 mL/min,进样量10 μL,柱温35℃,检测波长330 nm。
2.5.4 线性关系考察 精密吸0.5 mL、1 mL、3 mL、5 mL、7 mL、10 mL“5.1”项下的对照品溶液分别置10 mL容量瓶中,50%甲醇定容,摇匀,滤过,取续滤液按“5.3”项下色谱条件分别进样,以峰面积(A)为纵坐标,浓度(μg/mL)为横坐标,计算迷迭香酸的回归方程为Y=28251X-15.812(r=0.9996),说明迷迭香酸在118.72~4.24 μg/mL的浓度范围内线性关系良好。对照品及供试品的高效液相色谱图。如图4所示。

图4 迷迭香酸对照品(A)与肾茶饮片供试品(B)的HPLC图谱
2.5.5 精密度考察 取“5.4”项下同一浓度对照品溶液,按“5.3”项下色谱条件连续进样6次,按峰面积计算迷迭香酸的相对准偏差(RSD)为0.46%,说明仪器精密度良好。
2.5.6 重复性试验 取同一批肾茶饮片样品按“5.2”项下方法,平行制备6份供试品,按“5.3”项下色谱条件进样,按峰面积计算迷迭香酸RSD =0.66%,说明该方法重复性良好。
2.5.7 稳定性试验 取同一份供试品溶液,于0、2、4、8、12、24 h按“5.3”项下色谱条件进样,按峰面积迷迭香酸RSD =1.53%,说明供试品溶液在24 h内稳定性良好。
2.5.8 加样回收试验 取已知迷迭香酸含量的肾茶饮片样品,分别加入定量的迷迭香酸对照品,按“5.2”项下方法平行制备6份供试品,按“5.3”项下色谱方法进样,计算迷迭香酸的平均回收率为99.32%,RSD=1.61%,说明供试品的制备方法能将肾茶饮片中的迷迭香酸提取完全。
2.5.9 样品含量测定 按“5.2”项下方法制备各批肾茶饮片的供试品溶液,根据“5.3”项下色谱条件进样,计算其迷迭香酸的含量。结果表明,10批肾茶饮片的迷迭香酸的含量为1.05%~0.05%。具体见表3。鉴于SC10样品的迷迭香酸含量过低,舍弃,故按迷迭香酸含量下限值(0.19%)的80%制订限量标准,拟定肾茶饮片中迷迭香酸的含量不得低于0.16%。
表3 10批肾茶饮片中迷迭香酸的含量
3 讨论
迷迭香酸具有抑制和改善大鼠草酸钙型肾结石的作用[11-12],其与肾茶排石通淋的功效相一致,故本文以迷迭香酸为对照品进行薄层鉴别和含量测定。
本文在优选薄层色谱展开剂的过程中,对石油醚/环己烷-乙酸乙酯、二氯甲烷/氯仿-甲醇、甲苯-乙酸乙酯等展开系统进行了考察。结果发现,当以甲苯-乙酸乙酯-甲酸(5∶3∶0.8)为展开剂时,薄层展开效果最好,迷迭香酸对照品的比移值适中,供试品斑点分离明显,故选此为展开剂进行薄层鉴别。
为优选含量测定过程中最佳的供试品制备方法,本文对提取溶剂、料液比、提取方法、提取时间和提取次数进行考察。结果发现,100倍量50%甲醇超声提取肾茶饮片0.5 h即可将其迷迭香酸提取完全,故选此法制备供试品。
本文以不同比例的甲醇(乙腈)-水(磷酸水)为流动相,优选肾茶饮片中迷迭香酸的HPLC条件。结果发现,以乙腈-0.1%磷酸水(25∶75)为流动相时,供试品中迷迭香酸的分离效果好(R>1.5)且出峰时间(tR=10.10 min)适中,故选此为流动相测定肾茶饮片的迷迭香酸。
4 结论
本实验分别对肾茶饮片的性状、显微、薄层鉴别及水分、灰分、浸出物、迷迭香酸含量进行测定,为完善肾茶饮片的质量标准提供了实验依据。本实验所建立的关于肾茶饮片的定性和定量研究方法稳定、可靠,也为完善肾茶饮片质量标准提供了参考。
猜你喜欢
天津药学(2022年4期)2022-09-14
药学实践杂志(2022年3期)2022-05-27
中草药(2022年10期)2022-05-24
农业科技与信息(2021年16期)2021-09-10
健康之家(2021年19期)2021-05-23
中国药学药品知识仓库(2021年18期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
北方药学(2020年6期)2020-07-08
农家科技下旬刊(2018年2期)2018-06-24
绿色科技(2016年24期)2017-03-30
