
稠密流体氮高温高压相变及物态方程*
2022-08-12 14:28:22田春玲刘海燕王彪刘福生甘云丹
物理学报 2022年15期
田春玲 刘海燕 王彪 刘福生 甘云丹
1) (西南大学物理科学与技术学院,重庆 400715)
2) (西南交通大学,高温高压物理研究所,成都 610031)
3) (西安近代化学研究所,西安 710065)
氮的高温高压物态方程以及相图对于研究和制备高能量密度含能材料至关重要.本文采用基于密度泛函理论的分子动力学模拟方法,研究了液氮的高温高压行为,给出 900—25000 K,2—200 GPa 区间流体氮的物态方程以及组分、相态变化.在上述相空间,观察到流体氮分子相-聚合物相以及聚合物-原子相的相变发生.获得的液氮Hugoniot 理论曲线与实验结果吻合较好,发现30—60 GPa 区间Hugoniot 曲线的软化与分子-聚合物流体相的相变有关;在60 GPa 后Hugoniot 曲线变陡峭与流体氮进入聚合物相区有关.
1 引言
氮的高温高压行为一直受到人们的关注,不仅因为全氮化合物有望成为下一代的绿色高能量密度材料,也由于氮气是炸药主要反应和爆轰产物,人们在评估含能材料的能量特性和作功能力时需要了解氮的高温高压物态方程和相图[1,2].氮的最外层电子分布为2s22p3,在常温常压下氮原子与另外一个原子键合,形成配位数为1 的双原子分子化合物.氮分子中的氮三键(N≡N)是自然界最稳定的化学键之一,完全打破一个气相氮分子的N≡N 需要9.74 eV 能量[3].但在高压条件下,随着氮分子间距变短,分子间排斥力增大,分子内电子不再局域于分子内部,N≡N 变得不稳定[4];并随着温度的升高,N≡N 将断裂并形成单(N—N)、双键(N=N)链接的多原子分子化合物,即聚合氮[2,5−11].2004 年德国Max Plack 研究所Eremets 等[12]首先在2000 K 以及110 GPa 条件下观察到固氮从双原子分子晶体转变为单键原子构成的立方偏转氮(又称cg-N)结构,即1 个氮原子可与另外3 个氮原子相连形成3 配位的共价键化合物.最近,在静高压实验[13−16]中又相继在150 GPa,2000 K 下发现了一种层状聚合氮,该层状聚合氮与黑磷的晶体结构类似,具有褶皱蜂窝层状结构.此外,通过第一性原理计算人们预言了以单、双键链接的N8[10],N10[17,18]等分子形式存在的低压聚合氮晶体.在固态氮的高温高压相变研究如火如荼进行时,流体氮的高温高压行为也受到密切关注.
流体氮最早的研究工作可追溯到20 世纪70—90 年代,美国洛斯阿拉莫斯实验室Dick[19]首先通过炸药爆炸加载冲击波测量了液氮在2—43 GPa区间的冲击压缩Hugoniot 线;随后利弗莫尔实验室[20,21]利用气炮加载冲击波技术将液氮冲击压缩到20—80 GPa,并测量其冲击温度,处于5000—15000 K 间.研究发现液氮的冲击Hugoniot 曲线在30—60 GPa 区间,相应冲击温度为 7000—12000 K时出现软化现象.最近,国内西南交通大学动高压实验研究小组报道了类似的冲击波压缩实验测量结果[22].文献[23,24]利用了流体变分理论分析认为,流体氮冲击Hugoniot 曲线软化现象与液氮在30 GPa 和6000 K 时发生分子相到原子相的转变有关.近年来随计算机性能逐步提高,开始采用从头算模拟方法来研究液氮在高温高压极端条件下的行为,包括密度泛函分子动力学模拟(density-functional theory based molecular dynamics,DFTMD)以及路径积分蒙特卡罗模拟(path integral Monte Carlo,PIMC).普遍认为在中、低温区间,无需考虑电子的激发效应,DFT 理论比较可靠[25−28].2016 年Driver 等[27]利用PIMC 和DFT-MD 两种方法对高温流体氮的性质进行了研究;他们认为密度泛函理论以及交换关联泛函在T<105K 对于氮的描述是合理的,但在更高温度下PIMC 方法更可靠.2000 年Kress 等[29]利用DFT-MD 较好地描述了液氮的冲击Hugoniot 曲线,认为Hugoniot曲线软化与液氮分子相到原子相的相变有关.Boates 研究小组[30,31]采用DFT-MD 计算液氮的部分温压相区的物态方程,提出6000 K 以下分子流体分子氮的离解产物是聚合氮流体,在更高温度则是原子氮流体.最近,研究发现氢的高温高压离解产物是寿命较短的氢原子团簇,并与原子氢(等离子体氢)有明显区别[32].本文在氮的模拟中同样发现了大量寿命较短氮团簇聚合物,与原子氮相比它们的分子构型以及电荷密度分布完全不同.在对模拟数据进一步分析后,发现流体聚合氮可能比人们认为的更加稳定,存在温压范围更广.而液氮冲击Hugoniot 曲线在30 GPa 和6000 K软化现象更有可能是发生了分子相到聚合相的相变,而不是氮分子完全离解进入了原子或等离子体相区.
2 研究方法
本文基于密度泛函理论框架下的分子动力学模拟方法,研究了氮在高温高压下的物态方程以及相态变化,包括氮的压强、内能、组分等随温度、密度的变化规律.模拟工作采用VASP (Viennaab initiosimulation package)程序[33]完成.模拟超胞含有64 个氮原子(Nt=64),选用的电子交换关联函数为广义梯度近似(general gradient approximate,GGA)下的PBE (Perdew-Burke-Ernzerhof)泛函[34,35].为了保证计算的效率和精确度,电子波函数采用截断能Ecut为1200 eV 的平面波基组,总能量收敛精度达到 10−5eV,受力的精度为 10−4eV .布里渊区的能量积分采用 1×1×1 的网格点设置.分子动力学模拟采用正则NVT 系综,选用Nosé-Hoover 算法[36]控制离子温度,离子运动方程的积分以0.5—1.5 fs 时间步长进行计算,模拟时长至少为3 ps.
本文每个数据点的热力学量采集都在体系到达热力学平衡后才进行.考虑到氮原子间最大成键距离为单键键长(1.46 Å,1 Å=0.1 nm)[2,37],本工作与前人研究一致,选择氮原子间的有效成键半径为1.5 Å[30],即在该距离内的原子将与参考原子成键.根据成键情况对流体氮的组分中不成键的孤立氮原子(n=1,配位数为0)、氮原子对(n=2,即配位数为1 的氮分子)以及配位数大于1 的团簇聚合物(n≥ 3)进行统计,并用(Nn) 记录各组分布居数,组分比Rn定义为体系中各组分的原子数量相对于原子总数的百分比(RnnNn/Nt).而氮分子的离解度(β)指的是构成离解了的分子流体占超胞中原子总数的比例,即β1−2N2/Nt.各组分数Nn、原子的配位数是对达到平衡态的数千步分子动力学模拟轨迹进行统计分析得到的平均值.
3 结果与讨论
3.1 流体氮的等容物态方程与分子-聚合物-原子的相变
图1 和图2 所示为流体氮在25000 K 范围内流体氮的两条等容线.图1 给出了1.75 g/cm3下流体氮的压强(图1(a))、内能(图1(b))、原子配位数(图1(c))、组分(图1(d))随温度的变化关系,以及在不同温度下原子径向分布函数(图1(e))以及分子动力学模拟中超胞原子位置分布的随机抽样(图1(f)—(h)).从图1(a)可见压强P随温度T变化可以明显分为3 段;在2000—7000 K 范围,压强随温度单调增大;在7000—10000 K 时,压强随温度升高而下降;此后压强再次随温度升高而增大.
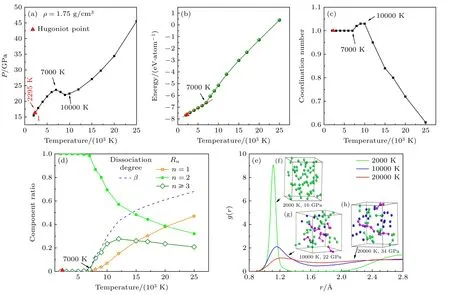
图11.75 g/cm3 时流体氮的等容线 (a)压强-温度关系;(b) 内能-温度关系;(c)原子平均配位数随温度的变化;(d) 化学组分占比(Rn)与离解度(β)随温度的变化;(e) 径向分布函数图像;(f)—(h)不同温度下的原子分布抽样,绿色原子代表氮分子组分,玫色表示聚合物组分,蓝色小球表示氮原子组分,红色三角形代表液氮的Hugoniot 状态点Fig.1.Fluid nitrogen at 1.75 g/cm3:(a) Temperature-dependence for pressure ;(b) temperature-dependence for internal energy;(c) temperature-dependence for coordination number of atoms;(d) temperature-dependence of chemical component ratio (Rn) and molecular dissociation degree (β) ;(e) pair correlation functions at temperatures of 7000,10000 and 20000 K;(f)–(h) snapshots from MD simulations.Green atoms represent molecules,the red spheres show polymers and the blue indicate isolated atoms.The red triangle represent the Hugoniot point.
在T<7000 K 相应P<24 GPa 时,氮原子的平均配位数(图1(c))保持在1,从图1(d)组分分析可见氮分子离解度为0,说明此时体系处于分子流体相.在7000 K≤T<10000 K 时,氮分子离解度快速增大,原子的平均配位数变得大于1,表明体系发生分子-聚合物相变,而非分子-原子相变.氮分子的垮塌造成等容线上压强负斜率变化((∂P/∂T)ρ <0),而内能则随温度的升高快速增大,斜率变大.组分分析也表明此区间氮分子快速变少,而聚合物较原子组分大幅增大,这揭示出氮分子解离后产生的原子更倾向与剩余分子结合,形成枝链状团簇(原子数目n≥ 3),如图1(g)所示,而形成孤立原子组分(n=1)的较少;体系是分子、大分子团簇为主的混合物.在T>10000 K 温度区间氮分子离解速率变缓,以至于分子离解对压强的负贡献可以被热效应的正贡献相抵消,故压强再次随温度的升高而增大.组分分析可见,聚合物产物在12000 K时占比最大,随后逐步减少,而原子组分则随温度的升高而稳步增加.在温度到达15000 K 的后,巨大的热运动动能使得原子间化学键极易短裂,组分中聚合物和剩余分子都将进一步分解,孤立原子逐步变成体系主要成分,原子的平均配位数小于1.如20000 K 下流体氮中原子组分占比超过40%,原子的平均配位数也只有0.7 左右,分子动力学模拟中超胞原子分布的随机抽样见图1(h),此时体系逐步过渡到原子为主的流体相.在流体氮从分子-聚合物的转变中,径向分布函数(图1(d))的第一峰的位置从7000 K 情形下的1.10 Å(对应双原子氮分子的N≡N 键长[30])移动到10000 K 下的1.15 Å,表明氮原子间键长变长,有N=N(键长1.25 Å[38])、N—N(键长1.46 Å)生成;在20000 K,34 GPa 的时,径向分布函数的峰值几乎完全消失,说明随温度升高体系逐步由聚合物向原子相转变.在聚合物-原子转变中,压强连续地随温度增大.
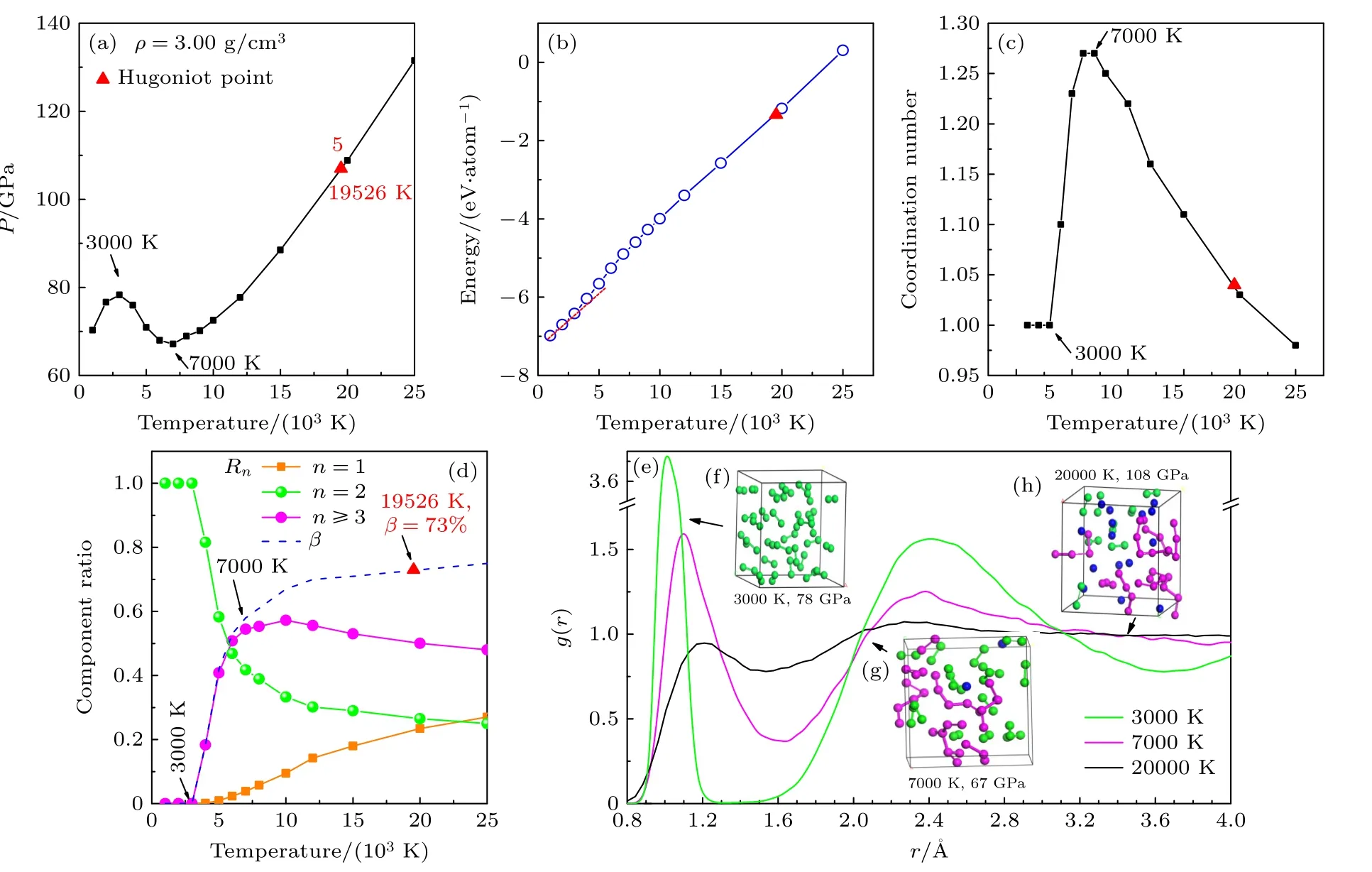
图23.00 g/cm3 时流体氮的等容线 (a)压强-温度关系;(b) 内能-温度关系;(c)原子平均配位数随温度的变化;(d) 化学组分占比Rn 与离解度β 随温度的变化;(e) 径向分布函数;(f)—(h) 不同温度下的模拟原胞原子的瞬态抽样Fig.2.Nitrogen isochore at 3.00 g/cm3:(a) Temperature-dependence for pressure ;(b) temperature-dependence for internal energy;(c) temperature-dependence for coordination number of atoms;(d) temperature-dependence of chemical component ratio (Rn) and molecular dissociation degree (β) ;(e) pair correlation functions at temperatures of 3000,70000 and 20000 K;(f)–(h) snapshots from MD simulations at different temperatures.
类似的流体氮从分子-聚合物的转化造成等容线上压强负斜率变化现象在3.00 g/cm3时也观察到,如图2(a)所示.随着密度变大,流体氮分子-聚合物相变发生在较低的3000—7000 K 温度区间,相变前后超胞原子瞬态分布如图2(f)和图2(g)所示;此区间压强随温度增大而降低,内能随温度的升高而增大且斜率变陡(图2(b)),体系原子的平均配位数从1 快速上升到1.28 左右(图2(c)),分子离解度从0 增大到60%,聚合物成分从0 上升到58%,而原子组分不超过2%(图2(d)).在10000 K左右聚合物组分含量达到峰值,随后逐步减少,而原子组分一直随温度的升高而增大.随温度的升高,径向分布函数的第一峰值逐步降低,位置从1.1 Å向1.3 Å移动(图2(e)).与1.75 g/cm3情形相比,3.00 g/cm3流体氮中孤立原子组分随温度增加极为缓慢;即便在20000 K 的高温下,热效应仍不足以使大部分原子克服化学键的束缚,变为自由原子为主的流体相;体系组分还是以聚合物居多(52%),原子组分虽然上升到22%,但仍不占优,如图2(h)所示;即体系仍处于聚合物为主相区,仍未进入原子流体相区.可见,密度(压强)越大,聚合物-原子流体的相变的温度越高.
当密度为1.75,3.00 g/cm3时,对流体氮在温度分别为2000 K,7000 K,20000 K 下的分子动力学模拟中超胞(即图1(f),(h)和图2(g),(h))的电荷密度进行计算,结果见图3.从图3 可以看出体系中原子的成键情况.图3(a)显示2000 K 以及1.75 g/cm3下流体氮中双原子成键,表明体系处于分子相;而图3(b)则显示20000 K 以及1.75 g/cm3下大多数原子的价电子局域在原子核周围,成球形,原子间无化学键,体系处于以原子为主流体相;图3(c)和图3(d)显示3.00 g/cm3时在7000 K 和20000 K时,体系除了原子、分子外,还有多原子成键的N3,N4,N5等大分子团簇存在,并在数量上占优,体系处于以聚合物为主流体相.可见,体系的电荷分布信息也支持流体氮处于分子相、原子相或聚合物相态的判断结论.

图3 在不同的温度和密度下流体氮结构的价电荷密度(a) 1.75 g/cm3,2000 K;(b) 1.75 g/cm3,20000 K;(c) 3.00 g/cm3,7000 K;(d) 3.00 g/cm3,20000 KFig.3.Valance charge densities of fluid nitrogen at different temperatures and densities:(a) 1.75 g/cm3,2000 K;(b) 1.75 g/cm3,20000 K;(c) 3.00 g/cm3,7000 K;(d) 3.00 g/cm3,20000 K.
3.2 Hugoniot 物态方程
在冲击波压缩下,波后液氮的内能、压强、密度满足Hugoniot 关系式:

其中,E0,P0,ρ0分别是液氮初始状态的内能、压强和密度.本文依照液氮Hugoniot 实验[19−22]对初始条件为T0=77 K,ρ0=0.808 g/cm3,P0=1 atm(1 atm=101.325 kPa)液氮的冲击压缩状态进行了计算,结果见表1,并把理论计算结果与测量结果进行比较,见图4(a)和图4(b).

表1 DFT-MD 模拟得到的液氮冲击状态数据以及流体组分(初态ρ0=0.808 g/cm3,T0=77.6 K,E0=–8.319 eV/atom)Table 1.Calculated results for pressure and temperature and the chemical components along the Hugoniot curve from our DFT-MD simulations.The initial conditions are ρ0=0.808 g/cm3,T0=77.6 K,E0=–8.319 eV/atom.
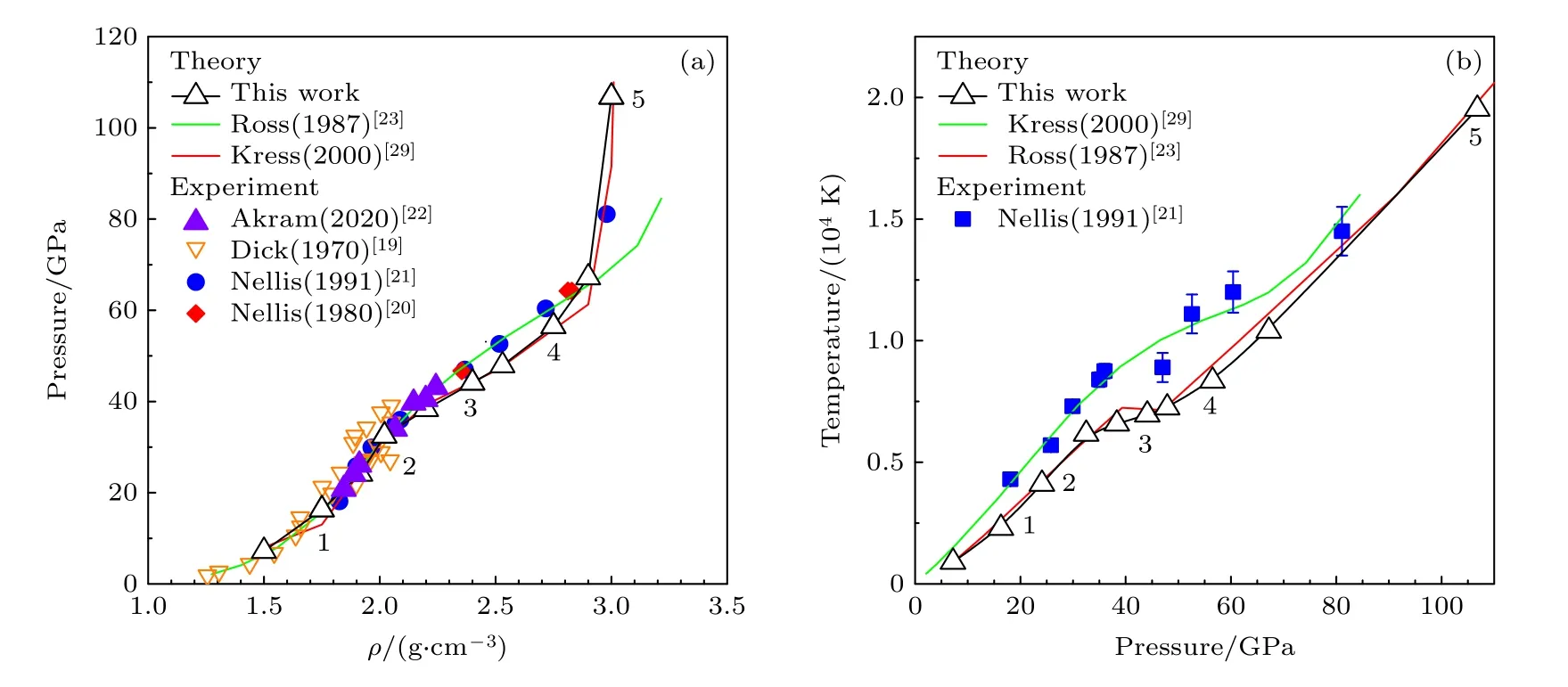
图4 液氮的Hugoniot 物态方程的实验和理论结果比较 (a)压强-密度关系;(b)温度-压强关系Fig.4.Comparison between the experiments and calculations of Hugoniot equation of state for liquid nitrogen:(a) Pressure as a function of the final shock density;(b) shock temperature as a function of the pressure.
本文计算得到的液氮Hugoniot 冲击压缩线与实验测量结果[19−22]符合得较好(图4(a)),在30—60 GPa 区间Hugoniot 线出现软化现象,在高于60 GPa 压力区间Hugoniot 线再次变陡峭.在Hugoniot 线出现软化时,冲击温度随压强上升趋势也变缓(图4(b)).图4(b)给出此区间的温度理论估值在6000—10000 K,略低于实验值7000—12000 K,同时给出了Kress 等[29]的从头算结果以及Ross 的经验模型计算结果进行比较.对于冲击温度,本文计算结果与Kress 等[29]的从头算结果均略低于实验值,但整体趋势与实验是一致的.Ross[23]的经验模型对冲击温度描述和实验结果较吻合,但Ross[23]的经验模型对Hugoniot 线在压强超过60 GPa 变硬的趋势很难描述.
为了对液氮Hugoniot 物态方程的变化特征做出解释,对Hugoniot 线上的5 个状态(在表1 和图4 标号为1,2,3,4,5)点进行分析,它们的密度分别为1.75,2.02,2.40,2.75 和3.00 g/cm3.这些冲击压缩状态分别处于相应等容线斜率为正的分子相区,斜率为负的分子-聚合物相变区以及斜率为正的聚合物相区.其中,冲击压缩密度为1.75 g/cm3状态1 的压强为16.3 GPa,冲击温度为2295 K(见图1 红色),它位于等容线的分子相区,体系离解度为0.冲击压缩密度为2.02 g/cm3的末态2,对应冲击状态(33 GPa,6164 K)刚好进入等容线的负斜率区间(如图5(a)所示),体系原子分布抽样如图5(b)所示;此时体系离解度为3%,表明分子-聚合物相变刚开始.冲击压缩密度为2.40 g/cm3的末态3 正处于等容线的负斜率区(如图5(c)所示),即体系处于分子-聚合物相变区,对应冲击压强约为44 GPa,冲击温度理论值为6955 K;此时约有27%的分子离解,并且产物绝大多数为聚合物(25%),体系原子分布抽样如图5(d).当冲击压缩密度到达2.75 g/cm3时,对应状态4 (如图5(e)所示),相应的冲击压强约为57 GPa,冲击温度计算值为8348 K,该状态处于等容线负斜率区的终点附近;此时过半的分子(53%)已离解,绝大多数产物是聚合物(47%),见图5(f)和图5(g);意味体系跨过分子-聚合物相变区间,刚进入聚合物相区.在3.00 g/cm3的冲击状态5 (107 GPa,19526 K)处于等容线的正斜率相区((∂P/∂T)ρ >0,如图2红色所示),其中聚合物占51%,原子只占22%,剩余是分子,体系组分构型抽样如图2(h),意味体系处于聚合物为主的流体相.
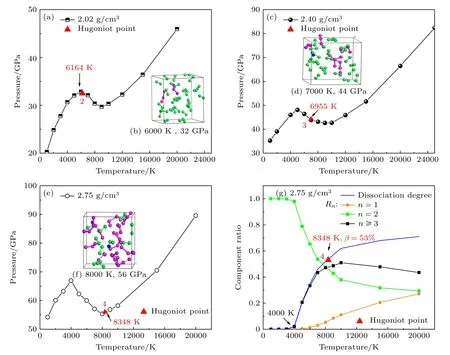
图5 液氮的Hugoniot 冲击 压缩点在不 同等 容线上的压 强-温度相空 间分布 (a) 2.02 g/cm3;(c) 2.40 g/cm3;(e) 2.75 g/cm3;图(b),(d),(f)是与图(a),(c),(e)相对应的分子动力学模拟中原胞原子位置分布的的瞬态抽样.(g) 2.75 g/cm3 下流体氮的组分Fig.5.Hugoniot points in P/T space at constant densities:(a) 2.02 g/cm3;(c) 2.40 g/cm3;(e) 2.75 g/cm3;(b),(d),(f) corresponding snapshots from MD simulations of panels (a),(c),(e).(g) Chemical components at 2.75 g/cm3.
理论计算结果表明,在状态2 和4 之间的30—60 GPa 冲击压强区域,体系处于分子-聚合物的相变区,由于分子的垮塌导致压强减小,因此实验上会观测到Hugoniot 线软化现象,并且得到的格林乃森参量为负().在该区域由于体系吸收的冲击波能量相当部分将用于打破分子键能的束缚,导致冲击温度(热效应)升高缓慢.在冲击状态4 (P≈60 GPa)时,体系完成分子-聚合物相变.此后,流体氮进入聚合物相区,体系冲击压强和冲击温度快速上升,Hugoniot 线变陡峭.本文理论模拟完美地解释了Hugoniot 线30—60 GPa 区间软化现象以及P>60 GPa 后变陡峭的测量结果.研究表明实验观察到液氮Hugoniot线的软化与流体氮的分子-聚合物相变有关,而非分子-原子流体相变导致,而聚合氮存在温压相区远高于目前认为的6000 K 的温度下.对于冲击温度,本文理论结果略低于实验值,可能原因是选用的交换相关泛函没有考虑色散相互作用而导致较多的分子离解,因为最近有研究[28]表明范德瓦耳斯相互作用将增加氮分子的稳定性.
4 结论
本文采用DFT-MD 方法研究了高温高压条件下氮的键合方式、组分演化与相态变化及其对热力学性质的影响.结果表明液氮分子相-聚合相的相变将导致等容线上压强随温度负增长变化.研究发现在30—60 GPa 区间Hugoniot 线的软化由液氮分子相-聚合相相变所致;在冲击压强高于60 GPa时,流体氮进入聚合氮为主相区,Hugoniot 线变硬.在目前实验所能达到20000 K 以及100 GPa冲击压缩状态下,原子氮流体组分只占20%,聚合物占51%,体系仍处于聚合物为主流体相,而非原子流体相.本文计算结果较好地解释了Hugoniot物态方程测量结果,表明聚合物流体相可能存在温压相区更广,远超过目前认为6000 K 温度范围.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
皮肤病与性病(2021年3期)2021-07-30 08:08:12
黑龙江动物繁殖(2020年2期)2020-12-17 14:39:26
同煤科技(2019年1期)2019-05-16 01:46:22
化工管理(2017年12期)2017-05-12 08:38:28
中国医疗美容(2015年2期)2015-07-19 10:11:59
中国畜牧兽医文摘(2015年4期)2015-01-24 21:31:02
动力工程学报(2013年3期)2013-08-16 00:23:10
