
光催化-生物杂合系统设计优化用于燃料和化学品绿色合成
2022-08-10 09:49张劢田瑶郭之旗王叶窦广进宋浩
化工学报 2022年7期
张劢,田瑶,郭之旗,王叶,窦广进,宋浩
(1天津大学化工学院,天津 300072; 2天津大学合成生物学前沿科学中心和系统生物工程教育部重点实验室,天津 300072)
引 言
近百年来,随着工业进程的高速发展,不可再生的化石能源滥用严重,导致CO2等温室气体过度排放,同时引发了严重的能源危机和各种环境问题。为实现可持续发展,2015 年《巴黎协定》首次提出全球“碳中和”概念,截至目前已有超过120 个国家和地区提出了自己的碳中和达成目标。其中,将CO2转化为碳基化学品及能源物质,以及使用清洁高效能源替代化石能源成为重中之重[1-3]。
由于自身稳定的热力学与动力学性能,CO2的活化并不容易[4],目前工业常用的几种将CO2转化为其他碳化合物的方法,如电催化[5-9]、化学重整[10]等,反应条件一般比较苛刻,而且依靠电能的还原反应会导致额外的能量输入,如今的电能提供依旧比较依赖于火力发电,无法完全摆脱化石燃料的污染。氢气作为高效清洁的新型能源物质,近年来逐渐受到青睐。然而,目前制取氢气的三种主流方法:电解水制氢、工业副产氢和化石燃料制氢,过程同样需要大量的能源消耗并造成温室气体排放,尚不能满足绿色化学以及“碳中和”理念的要求。作为农业氮素来源的合成氨技术,存在同样问题。以Haber-Bosch法为代表的人工固氮途径在铁催化剂、高温(500~600℃)、高压(20~50 MPa)下才可实现N2的转化,此后改进的一系列方法,如Marnellos 等[11]发明的常压合成氨手段,也需将电解池温度加热到570℃,据统计,平均每生产1 t氨,排放约2 t二氧化碳气体[12]。因此,如何利用安全清洁、可持续再生的新能源实现重要化学品的合成成为了人们亟待解决的问题。
地球上的能量核心——太阳能,由于具有储量丰富、可再生、分布广泛、安全、清洁等独特优势,被国际公认为未来最具竞争性的能源之一。1976年研究人员首次发现了能直接利用光能,在温和条件下催化化学反应的半导体材料,并由此开辟出光催化领域,实现利用太阳能安全清洁、绿色可持续地合成化学品。近年来,随着纳米技术的迅猛发展,光催化材料在光能转换效率方面得到大幅度提升,已远远超过自然界的光合作用[13-14]。然而由于光催化材料本身的催化特性,传统无机/有机光催化仍然存在着反应特异性较差、产物谱较单一、产物多为低碳简单化合物等问题,不能满足现阶段对靶向高效生产长链高值化合物的需求[15-18]。随着自然界的进化,生物体进化出高效的生物酶分子。通过特定的二级及三级结构,酶分子可降低催化能垒,调节催化方向,完成温和、高效、高产物特异性的催化过程。与此同时,成千上万的酶分子,通过生物体的精密调控,组成一条条精巧的代谢途径,可精准高效地合成各种长链高值化合物[19-21],这是化学催化所不可比拟的。
基于此,将光催化和生物催化耦合的光催化-生物杂合系统,近年来逐渐成为研究热点。该系统利用光催化高效的光能转化性能,将光能转化为高能还原物质/电子,为生物催化提供能量和还原力,最终实现绿色、高效的化学品合成。根据生物催化载体的不同,光催化-生物杂合系统可分为光催化-生物酶杂合系统和光催化-微生物杂合系统两大类,本文以电子传递机制作为划分依据,分别对这两类系统的作用方式进行分类(图1),详细评述各体系中光生电子的传导方式,以及存在的优缺点和关键问题,最后对该领域提出了展望。

图1 光催化-生物杂合反应体系分类示意图Fig.1 Classification of photocatalysis-biological hybrid systems
1 光催化-生物酶杂合系统
光催化-生物酶杂合系统是将光催化和单酶/多酶催化系统耦合,利用太阳能为酶催化提供能量和还原力的系统。和光催化-微生物杂合系统相比,此系统理论上可将转化后的能量和还原力完全用于目标产品的合成,避免了维持微生物生存所需的能量损耗,光能转化效率更高。但由于缺乏微生物精巧的代谢网络调控,酶催化系统在合成复杂的多碳长链化合物方面有所欠缺,更适用于简单化合物的合成。近年来,光催化-生物酶杂合系统在制氢、合成氨和CO2合成低碳化学品等领域展现出了瞩目的优势[22-27]。
根据活性位点是否被完全包裹在酶内部,光催化向酶分子供给能量和还原力的途径分为两种:当活性中心裸露在酶分子外部时,光生电子可以直接被其接收继而参与氧化还原反应;与此相对的,若活性中心完全被酶分子包裹,无法实现直接注入的电子就需依赖辅因子作为介体激活氧化还原作用,这两种情况中后者更为常见。基于此,光催化-生物酶杂合系统可以分为辅因子介导的间接体系、直接电子传递体系以及耦合二者的混合型体系。
1.1 基于辅因子介导的间接反应体系
大多数氧化还原酶的活性位点被掩埋在酶分子内部,光催化剂产生的光生电子无法直接抵达被包裹住的活性中心,因此需要利用辅因子[如NAD(P)H]作为穿梭介体为氧化还原过程提供能量和还原力。然而光催化剂直接还原再生辅因子往往会部分生成无生物活性的二聚体[NAD(P)2]和1,6-NAD(P)H,导致反应效率很低。这主要由于光催化剂和NAD+之间的能带结构一般不相匹配[28]。因此基于辅因子介导的间接反应体系普遍加入助催化剂降低反应所需活化能,以实现具有生物活性的1, 4-NAD(P)H 的高选择性再生。在各种助催化剂中,最常用的为铑复合物{[Cp*Rh(bpy)H2O]2+},NAD(P)+的C3原子上的酰胺可与Rh金属中心实现配位作用从而进行NAD(P)H的高效还原[29]。
Baeg 等[30]将靛红-卟啉(IP)发色团与石墨烯(CCG)共价连接而成的光催化剂CCG-IP 与酶的级联反应耦合,构建出了被誉为“人工叶片”的CO2固定产甲醇的装置,光生电子经铑复合物的介导可实现NADH 的高效再生,而NADH 则被用于甲酸脱氢酶、甲醛脱氢酶以及醇脱氢酶构成的三酶级联反应,最终光照30 min,体系可获得11.21 μmol/L 的甲醇[图2(a)]。但这种光催化剂、铑复合物、NAD+与酶之间游离的体系会使电子传递效率下降,导致体系中的活性氧增多、反应产率降低等问题,利用局域效应等手段增强有序的电子传递成为了改善体系的有效手段。Song等[31]通过将铑复合物缀合到噻吩修饰的C3N4光催化剂上,构建了高度集成的光催化-生物酶杂合系统,光催化剂与铑复合物的紧密共轭增强光生电子的转移效率,NADH 再生速率可达9.33 μmol/(L·min),比起其他同类的游离体系增强了2.33 倍。另外模仿类囊体膜而将甲酸脱氢酶包封入金属-有机骨架结构(MOFs)中,这种隔室化方法不仅避免了光反应产生的活性氧造成的生物酶失活,而且采用的MAF-7 这种MOFs 具备的亲水性和pH 缓冲能力也为生物催化提供了良好的微环境,从而体系反应9 h,甲酸的产量为16.75 mmol/L,是均相反应产物的3.24倍[图2(b)]。Ji等[32]模拟植物叶绿体,构建了卟啉/ SiO2/ Cp * Rh(bpy)Cl 杂化纳米粒子(TCPP/SiO2/Rh HNPs)。具体来说,将光敏剂TCPP、NAD+与甲酸脱氢酶均束缚于以SiO2为骨架的纳米粒子内部,而亲水性的铑复合物则分布在纳米粒子外侧,如此构建出的高度集成的单位活性结构有效提高了电子传递效率,将NADH 的再生率从11%提高到75%,将甲酸产量从15 μmol 增加到100 μmol。另外,集成的杂化粒子结构可实现铑复合物、酶和NAD+的低成本简便回收,并在重复利用10个循环后仍能保留80%的催化活性,极大降低了应用成本[图2(c)]。

图2 辅因子介导的间接光催化-酶反应体系Fig.2 Indirect photocatalytic enzyme reaction system mediated by cofactor
1.2 基于直接电子传递的反应体系
虽然很多酶的活性中心都被包埋在内部,但是仍然还有部分含血红素[33]或含黄素[34]的酶,其活性中心暴露在表面,在一定条件下可以直接接收光生电子进行酶催化反应;另外,光生电子也可以通过注入酶远端的铁硫簇,继而经由酶内部铁硫簇等组成的电子传递链,最终转移到催化活性位点。目前很多氢化酶[35-36]、甲酸脱氢酶[37]和固氮酶[23]等重要生物酶,都已经被发现可以进行这样的直接电子传递作用。这种直接电子传递的作用体系虽然适用的酶较少、应用方面较窄,但避免使用昂贵的铑复合物可以减少转化为NAD(P)H 导致的能量损失,简化了反应体系的同时也可以降低反应成本。
为了使光生电子有效地注入酶的活性中心,需要将氧化还原电势匹配、无生物毒性的光催化剂,限制在酶的活性中心或远端接收电子的铁硫簇附近,一般在20 Å(1 Å=0.1 nm)内的电子可以实现有效传递[35],如此量子点与酶之间需要存在一种相互作用来缩近并稳定二者之间的距离,否则激发的光生电子会有极大的概率与空穴复合发生猝灭,或者无法传递到酶分子中而导致反应终止。目前的光催化剂-酶直接偶联的方法包括静电、疏水相互作用、共价修饰或特异性蛋白质-氨基酸结合等[38]。这几种方法互有利弊,具体来说,光催化剂与酶之间的静电吸附[24,39]作用广泛存在,在不对酶进行复杂工程改造的情况下,通过简单的静电结合作用,也可能实现高效的光生电子的向酶反应中心的注入。但由于并未专门针对酶中的氧化还原位点的临近区进行设计,反应效果具有随机性,若结合位点与电子接收位点距离过远,会导致电子传递损失甚至无传递作用,此外,除了会受周围离子环境的干扰[40],长时间的反应中,静电吸附作用会变得不稳定,最终导致反应中断。类似地,虽然碳材料以及多环芳香化合物等,可通过π-π 相互作用与酶分子中的疏水区偶联结合,但电子传递效果同样存在不确定性。共价修饰具有最强的结合作用力,例如由于二苯并环辛炔(DBCO)与叠氮化物反应可生成稳定的三唑,在分别利用DBCO 与叠氮化物进行修饰后,酶与光催化剂即可实现高选择性的结合[24]。此外,利用偶联剂(EDC/NHS)也能实现光催化剂与酶分子的共价连接,虽然这类方法具有可操作性强、应用范围广、作用稳定等优点,但由于进行了有机分子的修饰,可能导致酶活性降低甚至失活。通过合理的蛋白质设计改造,可在不影响酶活性的前提下,实现光催化剂与酶的位点特异性结合,最大限度地保证电子传递效率。一般来说,过渡金属可通过配位作用,与酶分子上修饰的His-tag 结合[41],虽然作用效果较稳定,且在蛋白质分子的3′或5′添加组氨酸并不会对酶的活性造成损伤,但同时意味着蛋白质的电子接收位点附近,至少需存在一个肽链的端点才能实现有效电子传递,应用存在局限性。此外,可通过将电子接收位点附近的氨基酸诱变为半胱氨酸,以巯基结合作用实现酶与金纳米团簇(AuNCs)、银纳米团簇(AgNCs)等金属催化剂的特异性结合[35]。虽然这类特异性结合设计可达到最理想的电子传递效率,但需要大量的前期准备工作,包括酶三维结构信息的采集与分析,以及合成生物学改造等。
Reisner 等[42]引入半人工光电极[43]的概念,构建了光电驱动二氧化碳固定的体系,具体来说,是将生物提取的光合系统Ⅱ与固定了光敏染料的半导体结合,并作为光阳极,接受光照并产生光生电子,电子经由外电路传到由二氧化钛与甲酸脱氢酶(FDH)构成的阴极,通过FDH 表面的FeS 簇传导到FDH 氧化还原催化中心,实现二氧化碳到甲酸转化。此体系绕过了自然界中的Z-Scheme 和卡尔文循环的限制,如:光采集效率低及1,5-二磷酸核酮糖羧化酶(RuBisCO)周转率低(1~10 s-1)等,提高了光激发电子的传递效率。虽然表面上看起来是利用电极催化,但其反应的能量来源仅为太阳能,这种光伏电催化(PV-EC)体系有别于传统的电催化,避免了火力发电过程中的污染问题,本质同单纯的光催化系统(PC)一样,都是理想的反应体系。在将体系进行了进一步改进后,他们实现了甲酸脱氢酶(FDH)与金属氧化物(TiO2)偶联完成的CO2的催化还原[图3(a)][37],石英晶体微天平(QCM)和衰减全反射红外光谱(ATR-IR)证实了FDH 与TiO2表面的高亲和力,使得不存在氧化还原介质的情况下,也能实现光驱动CO2向甲酸盐的转化,周转频率(TOF)约为11 s-1。
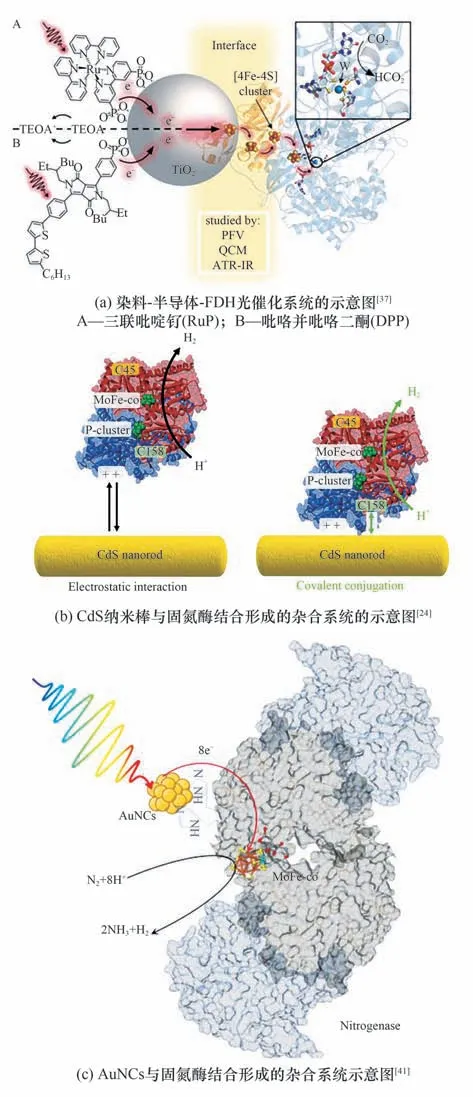
图3 直接电子传递的光催化-酶反应体系Fig.3 Photocatalytic enzyme reaction system based on directly electron transfer
King 等[44]研 究 了CdTe 纳 米 晶(NC-CdTe)和Clostridium acetobutylicum的[FeFe]-氢化酶I(H2ase)之间存在的静电结合以及电荷转移机制。经验证光生电子直接从核心纳米晶而不是表面陷阱态,转移到与NC-CdTe 对接的H2ase 远端[Fe4S4]团簇,再顺势传递到活性中心实现质子到H2的转化,在单色光下光子对H2的最高转化效率达到9%。Reisner 等[45]通过在CDs 表面修饰氨基得到的带正电荷的CDNMe2,可与[NiFeSe]-H2ase 表面,带负电的[Fe4S4]临近区实现静电结合,完成光激发电子向活性中心的传递,继而实现高效和稳定的底物转化与H2合成,而羧基修饰的、带负电荷的CDs(CD-CO2H),由于不具备结合作用,反应活性较低。Holá 等[40]通过研究天冬氨酸基碳点和[FeFe]氢化酶自组装而成的AspCD/CrHydA1 系统,揭示了牺牲电子供体形成的带电环境,对周围静电结合组件间相互作用的显著影响,具体来说,带负电荷的EDTA 可与CrHydA1 的表面正电口袋相互作用,从而妨碍CD(+)颗粒与酶之间的静电作用。而在TEOA 形成的良好的静电环境中,系统在420 nm 光照下,反应的外部量子效率可达到1.7%。Armstrong 等[35]通过将[NiFe]氢化酶中距远端[4Fe-4S]和内侧[3Fe-4S]10 Å 内的表面酪氨酸或苏氨酸改变为半胱氨酸,实现了银纳米簇(AgNCs)的位点选择性附着,继而完成光生电子供应氢化酶来驱动H2产生的过程。另外将形成的杂合体系负载在二氧化钛和石墨氮化碳(g-C3N4)构建的支架上形成的异质结构,能有效提高AgNCs 的光生电子-空穴的分离效率,每分子氢化酶每秒能产生40个H2分子。Harris 等[24]分别在CdS 纳米棒与MoFe 固氮酶上修饰巯基-聚乙二醇-叠氮化物和二苯并环辛炔-马来酰亚胺,继而实现二者的共价结合,大大提高了H2产量[图3(b)]。
Nagpal 等[41]研 究 了AuNCs 与His-tag 修 饰 的 固氮酶之间的电子传递作用,His-tag 位于MoFe 固氮酶α 亚基的C 端,与P 簇/FeMo 活性中心之间的距离很近,所以结合的AuNCs 激发产生的光生电子可直接注入到P 团簇,并转移到FeMo 中心,用于N2固定[图3(c)]。另外还研究了几种由于含不同金原子数,导致能带结构差异的AuNCs 的反应活性,结果表明Au22NCs 最适合与固氮酶偶联产NH3,而Au15和Au18等由于极低的VB位置并不利于生化反应。
除无机半导体外,有机配体、发色团以及共轭聚合物等也可应用于杂合生物酶体系[46]。生物酶的直接光激活可以消除其对辅因子与介质的需要,某种程度上实现了体系的简化,但同时也容易造成电子转移效率低和活性氧(ROS)产生等不利影响,关键就在于材料与酶之间的定向结合能否实现光生电子的有效转移,要精确地控制这种相互作用并非易事,而且酶的活性、三级结构也可能在进行化学修饰或与材料结合后发生改变。值得一提的是,在研究过程中CDs 由于具有廉价、低毒和可调节的表面化学修饰等优良性质,为人们广泛关注[47]。总地来说,将光催化和酶催化结合不仅可促进太阳能转化为燃料和增值化学品,也有望满足日益增长的绿色和可持续化学需求。
1.3 混合型光催化-生物酶杂合系统
铑基复合物等电子介体价格昂贵,且与生物催化剂相比周转频率(TOFs)低得多[48-50],此外,为达到必要的NAD(P)H 生产速率所需的相对较高浓度的铑配合物会导致生物质失活[51-52]。在自然界中,存在心肌黄酶(DH)、铁氧还蛋白-NADP+还原酶(FNR)[39,53-54]等一类含FAD/FMN 活性中心的酶,可以实现NAD(P)H 的合成,催化效率是铑基复合物的几倍[55-58]。同时,此类酶的活性位点有些直接暴露在表面,可直接结合光催化剂获得光生电子,高效率再生NAD(P)H。这种耦合了直接电子传递与辅因子介导两种作用而成的混合型光催化-生物酶杂合系统,同样继承了两方的优势,不仅应用面广泛、适用于活性中心暴露或不暴露两种类型的氧化还原酶,而且避免了铑复合物对生物酶的毒害作用、降低成本的同时提升了TOFs。截至目前,人 们 已 从 例 如Termotoga maritima[59]、Geobacillus stearothermophilus[60]、Bacillus subtilis[61]和Clostridium kluyveri[56]等微生物中,获得了不同种类的心肌黄酶。King 等[39]报道了利用硒化镉量子点(CdSe),和Fd(铁氧还蛋白)依赖的FNR 构建的杂合系统(图4),表面覆盖硫基丙酸(MPA)的量子点在Fd 结合位点上表现出特异性吸附。光照下NADP+还原为NADPH,平均TOF 为1440 h-1,在NADPH 作用下乙醛经醇脱氢酶催化实现到乙醇的转化,NADPH和乙醇的量子产量均为5%~6%。

图4 混合型光催化-生物酶杂合系统[39]Fig.4 A mixed photocatalysis-enzyme hybrid system[39]
1.4 维持生物酶活性的策略
由于光催化反应会产生大量的ROS,易导致酶失活,极大地限制了光催化-生物酶杂合系统的发展,所以如何在光反应中维持生物酶的活性成为了人们亟待解决的问题。加入适当的电子牺牲供体,以防止空穴氧化生成ROS 是最基本的手段,此外,促进高效的靶向电子传递作用,也是避免ROS 大量生成的途径之一。加入共/助催化剂(如CDs 或铑复合物等),可以促进对空穴或光生电子的捕获[62],从根源上减少ROS生成。另外,采用固定化酶手段,将游离酶包裹在MAF-7、ZIF-8 等MOFs材料中[31],将其与ROS隔绝,可在一定程度上保护生物酶活性。Jiang 等[63]制备了内部附有TiO2膜的SiO2涂层,并将醇脱氢酶(ADH)和光催化剂CdS 分别固定在其外表面和内表面,利用NAD+/NADH 在TiO2膜间的穿梭作用,实现了光催化与酶催化的隔室化反应,SiO2涂层可使ADH 免受光照伤害,并通过物理限制防止光催化产生的ROS 影响ADH 的活性。最后,通过额外加入过氧化氢酶(CAT)[64]、超氧化物歧化酶(SOD)等ROS 酶,实现ROS 的降解,一些具有类SOD、CAT 活性的物质如锰卟啉结构、铂纳米粒子等[65],也可被用于消除ROS,维持生物酶活性。
除了ROS,为尽可能维持生物酶在体外反应的活性,需要对缓冲液的类型[66]、光强、反应温度、压力等条件进行优化,并且还要考虑产物浓度对酶活性的影响。例如利用FDH 固定CO2合成甲酸时,过高的甲酸含量可能会导致pH 降低,影响酶活性。此外,体系中产物浓度的提高也会阻碍正反应的进行。因此,实现体系中产物的及时转移,即反应分离一体化,也是维持酶反应活性的策略之一。膜分离技术由于具有反应条件温和、无须引入其他物质等优势,不会对生物酶活性造成损伤,可用于酶发酵体系中产物的原位分离,装置可分为内置式与外置式两种,虽然可以在一定程度上解除产物浓度过高对酶活性造成的抑制,但是膜的高成本,与膜堵塞、污染等问题也会增加反应成本。除了提高酶活性,固定化技术也可用于酶的回收利用与连续发酵。通过包埋、吸附、共价结合、交联和亲和等作用[67],可将酶约束在反应器中并限制其自由流动,继而实现产物与酶的连续分离,保证酶在连续发酵过程中的活性不受产物浓度影响。
2 光催化-微生物杂合系统
光催化-微生物杂合系统是用微生物作为生物催化载体,利用太阳能为微生物代谢提供能量和还原力的系统。由于微生物具有精巧的代谢途径,就为合成各种高碳化合物提供了可能性,同时由于微生物具有一定的膜保护及自我调控能力,在体系抗逆性与稳定性方面相对酶体系更具有优势。与此同时,微生物为维持必要的生长代谢,需要消耗部分能量和还原力,导致光-产物转化效率降低。但在高值化学品的绿色合成方面,光催化-微生物杂合系统仍具有极大发展优势及潜力[68]。在合成生物可降解塑料PHB、长链脂肪酸等复杂化合物时,由于反应步骤烦琐,导致所需的酶种类过多,而目前多酶级联体系的构建依旧面临很大挑战,不仅存在酶的纯化工艺复杂、体外反应的稳定性较差等问题,产率也会随步骤增多而显著下降。但微生物由于内部已经进化出天然的代谢网络以及应激机制,一般情况下,选择合适的底盘菌就能得到完整的产物合成通路,且无须外部添加辅因子、ATP等生物反应的必需分子,而是由“细胞工厂”进行自发地合成及调配。
基于催化剂与微生物耦合方式的不同此系统可分为两大类,即胞外光催化剂的供能体系以及注入到胞质内的光催化剂供能体系,而前者根据电子传递机理的不同又可分为直接电子传递与化学物质介导两种。
2.1 基于直接电子传递的胞外能量供给模式
基于直接电子传递的微生物杂合系统实现的基础在于微生物表面存在能直接接收电子的物质[71-74],或是利用膜上氢化酶直接完成H2的生成,或是通过如铁氧还蛋白、黄素蛋白、细胞色素、OMCB等导电蛋白[75-77]或导电鞭毛等实现电子向胞内的引入。例如膜上含有导电蛋白的微生物包括希瓦氏菌(Shewanella)[78]、地杆菌(Geobacter)[79]等,可以实现电 子 跳 跃;而 鼠 孢 菌(Sporomusa)[80]、穆 尔 氏 菌(Moorella)[15]、梭菌属(Clostridium)[81]等,可通过膜上氢化酶接收电子直接实现氢气生成,而合成的氢气也可通过介导途径为胞内提供还原力[82];另外本身含有光合系统(PS)的微生物如红假单胞菌(Rhodopseudomonas)[63]则可通过PS 及电子传递链(PET)的作用接收外源电子,促进光合作用[83-85]。
Sakimoto 等[15]通过在非光合细菌Moorellathermoacetica表面自沉淀CdS纳米颗粒,使非光合微生物具有光敏性。在这个系统中,Moorella thermoacerica通过代谢半胱氨酸产生S2-,其与外加的Cd2+反应,实现了沉淀于微生物膜表面的CdS 纳米粒子的合成。在M. thermoacetica-CdS 杂合系统中,光生电子可以直接传递到M.thermoacetica的胞内并提供还原当量,利用Wood-Ljungdahl 途径来还原CO2生产乙酸,并随着微生物的繁殖,膜外CdS 纳米粒子也可进行再沉淀作用,表明体系具有动态组装及自我修复能力。他们后来又通过瞬态吸收(TA)光谱和时间分辨红外(TRIR)光谱,针对此杂合系统的电子传递过程进行了系统性研究[图5(a)][84],结果发现杂合体系的电子转移可能存在两种相互竞争的途径:直接注入到膜上可接收电子的受体蛋白如Fd、Fp等,实现ATP的合成并直接还原CH2-THF;而随着反应进程,氢化酶的表达量会增加,动力学显示电子更倾向于与膜上氢化酶或其他分子受体作用生成H2,再通过HydABC 复合物的作用进入Wood-Ljungdahl 途径。Qiao 等[86]针对此杂合系统的全局蛋白组和代谢物进行了详细研究,实验结果与之前提出的反应机理相符合,明确了光催化剂对微生物造成的代谢改变,Wood-Ljungdahl 途径被激活。与之类似的还有Jin 等[81]的研究结果,证明了Clostridium autoethanogenum与CdS 纳米颗粒形成的杂合系统,也可能同时存在电子向Rnf 杂合体(与NADH再生相关)以及氢化酶注入(后转到氢气介导作用)这两种直接电子转移的机制。
Wang 等[19]依照同样的自沉淀原理构建了CdS-Rhodopseudomonas palustris杂合系统,以实现光催化还原CO2合成生物可降解塑料——聚β-羟基丁酸酯(PHB)。其中光合系统(PS)可直接接收光能,产生的光生电子在电子传递链(PET)以及ATP 合成酶的作用下,为体系提供NADPH 以及ATP。而CdS 产生的光生电子则可通过PET 实现向细胞内的直接注入,进一步促进还原力再生,更有效地为细菌细胞内的CBB 循环供能,合成的3-磷酸甘油醛通过代谢转化最终生成PHB,产量较R. palustris提高了47%。另外此体系也可同时实现N2的固定[图5(b)],根据光合效率(PE)的计算,CdS 包覆的R. palustrisPE 为6.73%,比天然细胞(2.35%)高186%,优于许多光异养细菌[85],固氮酶与卡尔文循环同时工作时,体系固定N2和CO2的比例分别为71.9%和28.1%,表面包覆CdS的光诱导电子赋予生物杂交细胞更大的生存能力。

图5 基于直接电子传递的胞外供能模式Fig.5 Energy supply model based on directly electron transfer extracellularly
Fu 等[87]构建了一种采用n 型半导体TiO2纳米线阵列作为光阳极,与生物阴极联合的双室反应体系,利用太阳能作为唯一的能量输入,光生电子经由外电路直接传递给厌氧活性污泥中的微生物,最终实现CO2转化为CH4。利用循环伏安法检测阴极室过滤后的无细胞上清液,发现没有氧化还原峰且电流可以忽略不计,排除了体系中存在可溶性电子介体的可能性,验证了电子是从电极直接转移到微生物的作用方式,由此导致了96%的极高法拉第效率。
2.2 基于化学物质介导的胞外能量供给模式
合成生物学改造难度小、产物谱丰富的生产用工程菌株往往无法实现电子的直接跨膜传递,为了能够和更广泛的工程菌株进行适配,可以通过化学物质作为载体介导电子传递[88],突破电子传递模式只能用于亲电微生物的限制,实现更多高价值化合物的合成。常见的介导物质有甲基紫精、氢气与甲酸等。
甲基紫精(MV)是最早用作光催化-微生物杂合系统的电子介体,其氧化形式(MV2+)可以通过光催化作用还原为甲基紫精阳离子(MV·+),进而介导电子传递[89]。Honda 等[90-91]利用TiO2作光催化剂、甲基紫精(MV)为电子介体,实现了无贵金属的全细胞催化产氢作用,并通过改善反应条件优化了甲基紫精的再生效率,继而提高了光催化产氢率[图6(a)]。但体系需严格厌氧,否则电子无法传递给氢化酶进行氢气生成。而Zhao 等[92]通过仿生硅矿包裹构建了核壳结构的大肠杆菌聚集体,在空气条件下,SiO2壳中位于外层的、暴露在有氧条件下的菌体,将进行有氧发酵并逐渐消耗氧气,而内部的菌体则自然避免了氧气的干扰,能够实现在空气条件下的全细胞光催化产氢。利用膜结合的重金属螯合蛋白PbrR与镉离子结合,实现的CdS 纳米粒子的原位合成与表面展示,通过加强临近效应缩短了甲基紫精从CdS 处接收电子,到将电子传导入胞内[NiFe]氢化酶的距离,提高了电子传递效率以及产氢性能。值得注意的是,如CdS等含有硫族元素的光催化剂,由于易被光生空穴氧化,所以存在严重的光腐蚀问题。醇、胺、抗坏血酸和EDTA 等物质通常被作为牺牲还原剂,来清除光生空穴从而保持光催化剂的稳定性,而Zhao 等[92]就是通过添加100 mmol/L 的抗坏血酸,维持了系统的正常运转。

图6 基于化学物质介导的胞外供能模式Fig.6 Energy supply model based on chemical-mediated extracellularly
甲基紫精虽然有一定的应用前景,但是它们对微生物的毒害性较大。而氢气作为清洁无害物质也同样可以成为电子载体,在膜结合氢化酶的作用下可分解为菌体提供ATP,与此同时可溶性氢化酶也可以利用氢气产生胞内还原力NADH。Zhang 等[64]在以TEOA 为电子供体的情况下,采用光催化剂g-C3N4收集光能产生氢气,而氢气作为电子载体,分别在膜结合的氢化酶(MBH)与可溶性氢化酶(SH)的作用下,为Ralstonia eutrophaH16 提供ATP 与NAD(P)H,最终将PHB 产量提升至1.4 倍。经过后续研究,将电子供体从对微生物有毒性的TEOA 优化为水,提高生物相容性的同时也使体系更趋近于自然光合作用[93]。同时g-C3N4-过氧化氢酶-R. eutropha这一改进体系,通过过氧化氢酶,将材料表面由水分子捕获价带空穴而积累的H2O2分解为H2O 与O2,后者与H2均为微生物气体发酵提供基础,同时避免了光催化生成的H2O2对微生物造成的损害[图6(b)]。此光催化系统具有优异的光能到化学能转化效率,氢气产量可达55.72 mmol/h,经48 h 光发酵PHB 的产量达到41.02 mg/L。这种纯粹的氢气介导机制中,体系中的氢气是通过光催化材料实现的生产,而非光生电子向微生物膜上氢化酶注入而得的,与前所述的体系相比这是一个根本性的不同点。
甲酸也是一种常见的电子载体[94],其一定程度上解决了H2由于过低的水溶解度导致的传质效率和电子传递速率过低的限制,甲酸在膜结合的甲酸脱氢酶与可溶性甲酸脱氢酶的作用下,可分别为菌体提供ATP 与NAD(P)H,同时分解而成的CO2可直接参与CBB 固碳循环。Song 等[95]通过甲酸脱氢酶(Cs-FDH)实现CO2到甲酸的胞外转化,而甲酸此时作为碳载体与电子载体进入细胞,可为细胞提供还原力与碳来源。最后通过异源表达1,5-二磷酸核酮糖羧化酶(Rubisco)提高固碳效率,能使CO2更有效地实现到PHB 的生物合成,120 h 内产量达到了485 mg/L。
虽然间接介导的电子转移模式一定程度上减少了直接电子传递模式对微生物提出的必要限制因素,但从电子转移效率来看,前者在物质转化的过程中不可避免地会导致额外的能量损失。
2.3 胞内的能量供给模式
合成代谢过程基本在细胞质中进行,胞外的电子与氧化还原穿梭分子通过膜向细胞质的传递过程不仅会消耗额外的能量,同时也受到膜扩散的限制[98]。为解决这一问题,Yang 等[96]提出了一个新的光催化-生物杂合系统组装策略,将光催化材料导入到菌体内部,在胞内直接实现还原力与能量的供给。他们将生物相容性及稳定性良好的金纳米团簇(AuNCs)导入到M. thermoacetica内部,AuNCs 直接在胞内吸收光能,产生的光生电子可以直接转移到酶及其他介质上实现还原力的再生[图7(a)]。用结构照明显微镜(SIM)及能量色散X 射线光谱(EDS)阐明了细胞摄取AuNCs的可行性及材料在胞内的稳定性。此外AuNCs 同时具有猝灭活性氧的作用,降低了材料对菌体的伤害,提高了生物相容性,AuNCs 也不存在之前CdS 体系中材料的光氧化问题,因此不仅提高了能量效率,提高了乙酸产量,也大大改善了光反应系统中的稳定性,使反应活性可以维持至少4 d。
注入到细胞质内的光催化剂会与酶进行随机结合,造成能量损失,而Ding等[97]实现了光催化纳米量子点与生物酶的胞内定向结合[图7(b)],这种直接电子传递机制提高了光生电子的利用效率,进一步实现了人工光合系统的优化。通过His-tag 修饰,A.vinelandii胞内固氮酶和R.eutropha膜上氢化酶分别可实现与CdS@ZnS QDs 的特异性结合,大大提高了NH3与H2产量,在1.6 mW/cm2的LED 灯照射下,借助膜上氢化酶及呼吸链反应产生的ATP,可使PHB 产量提高到野生型的150%,自然光下扩大的传统生物反应器的高产量表明了用太阳能实现大规模生产的可能性。Yong 等[98]通过将CuInS2/ZnS 量子点转移到表达周质氢酶的Shewanella oneidensis中,构建了一个独特的周质光敏生物杂交系统[图7(c)]。QDs光敏剂的光激发和电子转移过程同时发生在Shewanella oneidensis的周质中,缩短了电子输运的距离,避免了跨膜过程中产生的额外能量损失,此外狭窄的周质空间使局部氢化酶浓度提高,增加了酶和光敏剂之间相互作用的概率,光生电子通过氢化酶中的Fe-S簇迅速转移到活性中心,实现质子还原和H2的产生,产量是裸量子点的8.6倍。

图7 基于胞内量子点的能量供给模式Fig.7 Energy supply model based on intracellular quantum dots
将光催化纳米粒子注入胞内继而实现还原能供给的体系还处于起步阶段,电子传递机理尚不明确。与此同时,定向结合的模式虽然减弱了光生电子随机传导对微生物造成的危害,但仍无法避免空穴利用如谷胱甘肽、半胱氨酸等常见胞内还原物质,造成代谢布局紊乱的潜在可能性,将对材料的生物相容性提出更高要求。
3 总结与展望
基于酶与微生物的这两类杂合体系互有利弊[99]:前者反应简便快速,但生物酶不仅提取分离步骤烦琐,在体外也极易丧失活性,导致体系稳定性低,且简单的单酶反应无法实现低成本底物到高价值化合物的转化,而多酶级联体系的构建困难大,且转化率逐级降低;与此相对的,光催化-微生物杂合系统由于微生物可进行自体繁殖,加上各种膜结构以及应激机制的保护,其稳定性相较酶系统有极大程度的提高,且通过微生物体内代谢网络的调控,可实现多种复杂化合物的定向合成,但是体系存在一定的构建及调控难度[100]。
虽然光催化-生物杂合系统的构建还存在一些问题,但经过不断研究也已经初步探索出了一些可行的解决手段。材料方面,除了进行改性修饰改善电子-空穴复合率、通过整合互补技术提高太阳能利用率[101]外,重要的是要兼顾其生物相容性、光照下生物毒性、体系适配性等问题。通过外层包裹谷胱甘肽(GSH)或半胱氨酸(Cys)等配体,可显著降低材料的生物毒性[102]。由于材料与蛋白结合可能影响蛋白三级结构致使其失活,与膜的结合涉及到吸附、膜变形甚至颗粒的内吞封装,大颗粒可能会导致膜穿孔造成细胞死亡[103],在这一点上尺寸较小的量子点具有优势,而球形颗粒则被证明是微生物接受度最高的形态[104]。材料的能带结构也是影响其生物相容性的重要因素之一,过高或过低的能带位置会破坏生物体原本的代谢布局并产生大量ROS以至体系失活[38],由此选用光电性能与电子转移蛋白及各类辅因子相匹配的光催化剂,以及适当的电子供体尤为重要。另外,由于紫外线或更高能量的辐射光会导致酶失活、DNA 损伤和细胞死亡,材料的吸收范围最好调整为可见光,CdS 作为相对成熟的模式材料有一个缺陷就在于它依赖紫外区,而InP 作为一种吸收可见光的光催化材料或许未来能有广阔的发展[105]。
合成生物学技术的发展也为杂合系统的构建及应用提供了有力手段,通过蛋白质工程可以实现酶的定向改造,通过氨基酸突变以及其他修饰改造可实现材料与酶分子的定向结合[24],为促进电子向酶活性中心的注入,或实现酶固定化从而维持活性创造条件。另外,通过基因工程改造,可实现光催化剂与微生物之间自发的生物相容性组装,或光催化剂的原位生物合成[106-109]。如QDs 可在细胞内与经His-tag修饰的固氮酶或氢化酶实现特异性结合,继而激活光催化-微生物杂合系统,完成固氮或固碳作用[97]。而Zhong 等通过基因编辑,将大肠杆菌生物膜中的CsgA 蛋白与His-tag 融合表达,得到的菌株能与无机纳米材料实现动态自组装[110],而后通过表达具有矿化能力的短肽,设计合成的TcReceiver/CsgAA7生物膜,还可实现CdS NPs 在细胞外,高特异性、高生物相容性的原位合成,得到的稳定的生物膜具有光电响应,可被用于全细胞光催化反应[111]。此外,利用表面展示技术实现PbrR 在细胞膜上的表达[109],可提高细胞对重金属离子的亲和力,继而促进纳米粒子的生物合成,实现光催化-生物杂合系统的构建及应用。
最后,有关电子传递效率方面,如何实现材料与生物质之间的良好相互作用,以最大限度地提高电子转移效率也是至关重要的命题,虽然目前的很多研究旨在阐明杂合系统的机制,但仍然还有很多不确定性,全面且明确地掌握杂合系统作用原理是实现清洁高效生产燃料及增值化学品的重要基础,基于本质出发才能更好地实现对光催化剂、生物质的优势改造,促进物质和能量在无机-生物界面的传导,最终提高产品效益。
光催化-生物杂合系统具有可持续、高效产专一化学品的能力[105,112-114],无论从新能源开发还是固碳角度来说都是实现“碳中和”的重要手段,在进行温和、无污染的催化反应的同时,还能实现低成本底物到单一增值化学品、清洁能源的可持续高效转化[68],其潜在优势还表现在其他方面,如能量效率可远高于自然光合效率(<1%)[115],为提高反应效率、优化体系等提供了基础,是自然光合作用的复现、简化和优化,未来糅合仿生学理念可进行更深层次、更广泛性的研究,是人类利用自然、改造自然的合理举措。
猜你喜欢
科学之友(2022年11期)2022-11-03
内江科技(2022年9期)2022-10-27
辽宁石油化工大学学报(2021年6期)2022-01-04
运输经理世界(2021年17期)2021-04-28
种子(2021年3期)2021-04-12
无机盐工业(2020年1期)2020-12-31
物理化学学报(2019年2期)2019-12-24
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
山东工业技术(2016年21期)2016-11-24
