
一测多评法测定混合核苷片中核糖核苷含量
2022-08-09 04:50李婵温锦荣刘昭张国荣王震
中国药业 2022年15期
李婵,温锦荣,刘昭,张国荣,王震△
(1.陕西省药品和疫苗检查中心,陕西 西安 710065;2.西安万隆制药股份有限公司,陕西 西安 710119)
混合核苷为核糖核酸经麦芽根浸提液在酸性条件下进行酶解所得产物与玉米淀粉的混合物,含胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷等多种核苷。混合核苷片是以混合核苷为主药制成的片剂,临床用于急慢性肝炎、肝损伤及肝硬化的辅助治疗,也可用于因辐射及放射治疗或化学治疗引起的白细胞减少症和非特异性血小板减少症或白细胞减少症[1-2]。现行国家药品标准中采用紫外-可见分光光度法控制制剂中核苷混合物的总量[3],但检测范围广,在该波长处有吸收的物质均可被检出。在原标准基础上建立高效液相色谱法[4-8],采用一测多评法实现多成分的质量控制,同时解决对照品不易获得的情况,选定合适的内参物,根据制剂各成分间存在的线性或比例关系,建立内参物与其他成分间的相对校正因子[9-14],只需测定内参物的含量,通过校正因子计算其他成分的含量。为此,本研究中建立了鸟嘌呤核苷、胞嘧啶核苷、尿嘧啶核苷与内参物腺嘌呤核苷的相对校正因子,通过外标法测定腺嘌呤核苷的含量,根据腺嘌呤核苷与其他成分间的相对校正因子计算胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷的含量,并与外标法测定的4种核糖核苷含量结果进行比较。现报道如下。
1 仪器与试药
1.1 仪器
岛津LC-2030C Plus型高效液相色谱仪、岛津LC-2010CHT型高效液相色谱仪(日本岛津公司);Waters 2695 型高效液相色谱仪(美国Waters 公司);TDL-40B型台式离心机(上海安亭科学仪器厂);MS105DU 型电子天平(梅特勒-托利多仪器<上海>有限公司,精度为0.1 mg);CP225D 型电子天平(赛多利斯科学仪器<北京>有限公司,精度为0.1 mg)。
1.2 试药
腺苷对照品(中国食品药品检定研究院,批号为110879-201703,纯度为99.7%);鸟苷对照品(批号为T277G005,标定纯度为0.3%),胞苷对照品(批号为R088G005,标定纯度为99.4%),均购自德国CNW Technologies GmbH 公司;尿苷对照品(Sigma -Aldrich公司,批号为BCCC2099,标定纯度为100.0%);混合核苷片(西安万隆制药股份有限公司,国药准字H20057365,批号分别为181206,181207,181208,181209,181210,181211,200902,201001,201002,201003);甲醇(色谱纯,美国Fisher公司);蒸馏水(广州屈臣氏食品饮料有限公司)。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:迪马Platisil ODS 柱(250 mm × 4.6 mm,5µm);流动相:30%甲醇(A)-5%甲醇(B),梯度洗脱(程序见表1);流速:1.0 mL/min;检测波长:260 nm;柱温:30 ℃;进样量:20 µL。在此色谱条件下混合对照品溶液胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷保留时间的RSD分别为0.03%,0.08%,0.11%,0.05%,峰面积的RSD分别为0.06%,0.05%,0.09%,0.19%。4 种核苷保留时间的RSD均小于1.0%,峰面积的RSD均小于2.0%,表明系统适用性试验结果符合要求。胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷的理论板数分别为8 192,10 698,15 208,144 254;分离度分别为7.128,19.965,26.910。

表1 流动相梯度洗脱程序(%)Tab.1 Gradient elution procedure of the mobile phase(%)
2.2 溶液制备
供试品溶液:取样品20片,研细,取细粉适量(约相当于核苷混合物0.16 g),精密称定,置100 mL 容量瓶中,加水适量,超声处理10 min 使提取完全,用水稀释并定容,摇匀,离心(转速为3 500 r/ min)10 min,精密量取上清液5.0 mL,置50 mL容量瓶中,用水释,摇匀并定容,0.45µm滤膜滤过,取续滤液,即得。
混合对照品溶液:取胞嘧啶核苷对照品15 mg,精密称定,置100 mL容量瓶中,加水使溶解并定容,摇匀,作为胞嘧啶核苷对照品母液(质量浓度为0.15 mg/mL)。分别取尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷对照品11.5 mg,精密称定,分别置25 mL 容量瓶中,加水使溶解并定容,摇匀,作为尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷对照品母液(质量浓度均为0.46 mg/mL)。分别精密量取上述4 种核苷对照品母液各1.0 mL,置50 mL 容量瓶中,加水稀释并定容,摇匀,即得。
阴性对照品溶液:按处方比例制备不含混合核苷的阴性样品,按供试品溶液制备方法制备相应的阴性对照品溶液。
对照品定位溶液:精密量取上述4种核苷对照品母液各1.0 mL,分别置50 mL容量瓶中,加水稀释并定容,摇匀,即得。
2.3 方法学考察
专属性试验:取2.2 项下阴性对照品溶液、对照品定位溶液、混合对照品溶液、供试品溶液各适量,按2.1项下色谱条件进样测定,结果各成分与相邻组分分离较好。色谱图见图1。
线性关系考察、检测限与定量限确定:分别精密量取2.2 项下核苷对照品母液1.0 mL,置25 mL 容量瓶中,加水稀释并定容,摇匀,作为线性溶液6;分别精密量取上述溶液1,2,3,5,8 mL,置10 mL 容量瓶中,加水稀释并定容,摇匀,分别作为线性溶液1-5。按2.1项下色谱条件进样测定,以胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷质量浓度(X,µg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程,详见表2。取2.2项下对照品溶液,逐级稀释,以信噪比分别为10∶1和3∶1 的质量浓度计算定量限和检测限。按2.1 项下色谱条件进样测定。结果见表2。
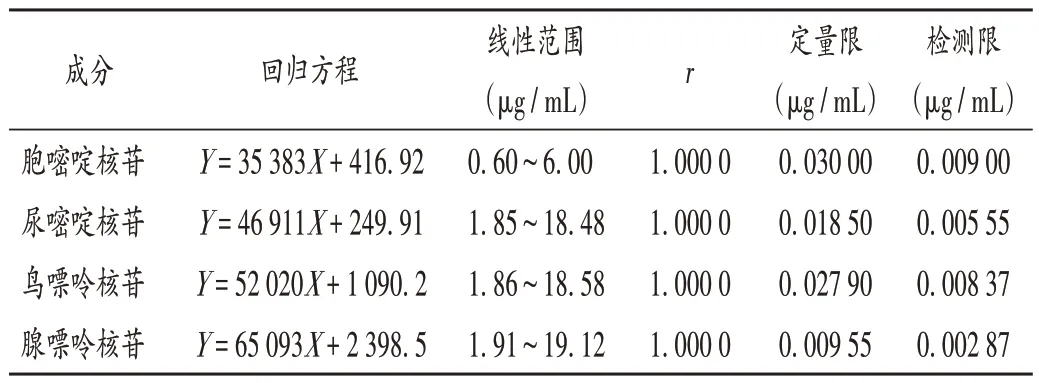
表2 线性关系考察、定量限与检测限确定结果Tab.2 Results of the linear relation test,LOQ and LOD
重复性试验:取样品(批号为201001)适量,精密称定,按2.2项下方法制备供试品溶液6份,按2.1项下色谱条件进样测定,记录各成分的峰面积,计算得胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷的平均含量分别为2.30%,6.03%,6.62%,9.10%,RSD分别为1.40%,0.63%,0.88%,0.44%(n=5),表明方法重复性良好。
精密度试验:取重复性试验项下供试品溶液6份,按2.1 项下色谱条件进样测定,记录各成分的峰面积,计算胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷的含量,平均含量分别为2.28%,6.12%,6.58%,9.01%,RSD分别为1.80%,1.60%,0.92%,1.10%(n=6),表明仪器精密度良好。
稳定性试验:取重复性试验项下供试品溶液,按2.1 项下色谱条件分别于0,2,5,9,12 h 时进样测定,记录各成分的峰面积,计算胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷峰面积的RSD分别为1.6%,0.58%,0.79%,0.50%(n=5),表明供试品溶液在12 h内稳定性良好。
加样回收试验:取胞嘧啶核苷对照品30 mg、鸟嘌呤核苷对照品75 mg、尿嘧啶核苷对照品75 mg、腺嘌呤核苷对照品115 mg,精密称定,分别置100 mL容量瓶中,加水溶解、稀释并定容,摇匀,作为各个成分的回收率对照品母液。取样品(批号为201001)细粉适量(约相当于核苷混合物0.08 g),精密称定,分别加入上述回收率对照品母液4,5,6 mL,按2.2项下方法制备供试品溶液。按2.1项下色谱条件进样测定,记录各成分的峰面积。计算得胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷的回收率分别为99.4%,99.1%,100.5%,99.9%,RSD分别为0.83%、0.28%,0.81%,0.63%(n=9),表明结果准确度良好。
耐用性试验:取2.2项下混合对照品溶液与供试品溶液,按2.1 项下色谱条件采用不同色谱仪、色谱柱进行检测,记录各成分的峰面积,按外标法以峰面积计算胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷的标示含量。与2.1 项下色谱条件比较,不同色谱仪和色谱柱所得胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷的标示含量的变化率分别为1.20%,0.25%,1.50%,0.23%,4 种成分标示含量总和的变化率为0.83%,均小于2.0%,表明方法耐用性良好。
2.4 相对校正因子(fs/i)
fs/i的建立:根据公式fs/i=(As×Ci)/(Ai×Cs)计算一测多评的fs/i[14]。式中,As代表内参物腺嘌呤核苷峰面积,Cs代表内参物腺嘌呤核苷质量浓度,Ai代表其他组分峰面积,Ci代表其他组分浓度。以腺嘌呤核苷为内参物,计算腺嘌呤核苷与胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷的fs/i分别为1.84,1.40,1.25,RSD分别为0.22%,1.75%,0.41%。结果见表3。
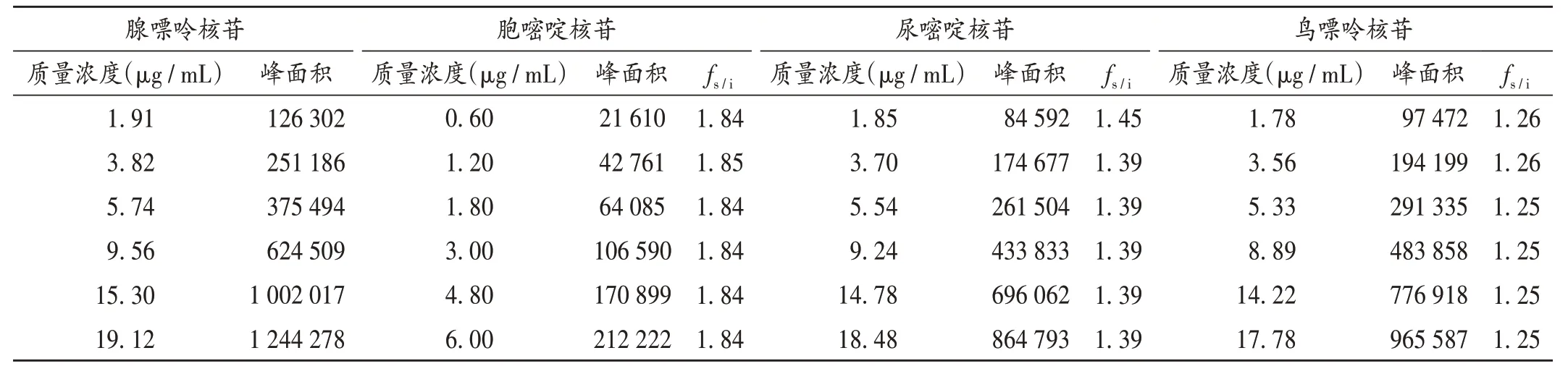
表3 胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷与腺嘌呤核苷的相对校正因子Tab.3 RCFs of cytidine,uridine and guanosine relative to adenosine
不同高效液相色谱系统和色谱柱对fs/i的影响:分别用Waters 2695、岛津LC -2030C Plus 2 种高效液相色谱系统进行试验,考察了迪马Platisil ODS 色谱柱(250 mm × 4.6 mm,5 µm)和InertsilTMODS -3 色谱柱(250 mm×4.6 mm,5µm)对fs/i的影响,各成分在不同色谱系统和不同色谱柱上fs/i的RSD均小于5.0%[15]。详见表4。
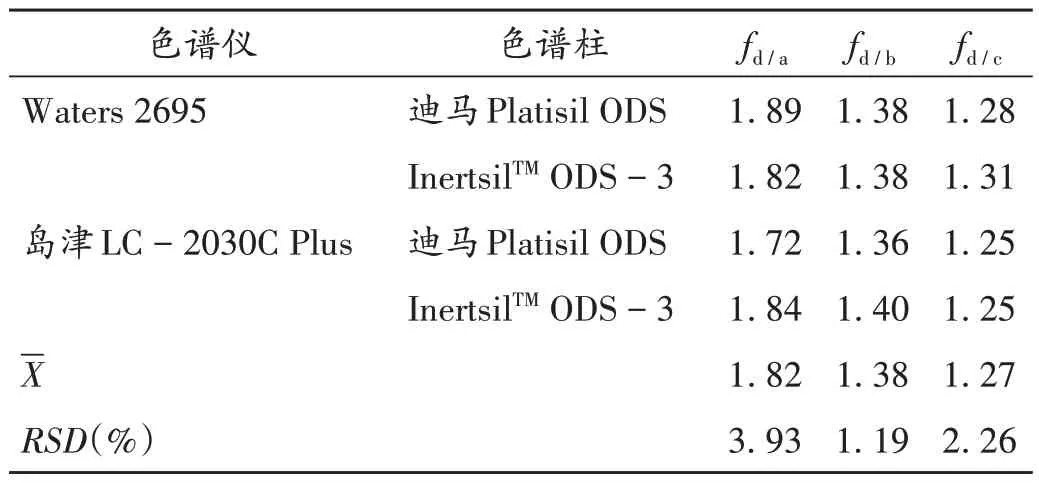
表4 不同色谱仪和色谱柱对相对校正因子的影响Tab.4 Effects of different chromatographs and chromatographic columns on the RCFs
2.5 一测多评法与外标法测定结果比较
取10 批样品,按2.2 项下方法制备供试品溶液,按2.1 项下色谱条件进样测定,测定各成分的峰面积。以腺嘌呤核苷为内参物,计算腺嘌呤核苷与其他3种成分的fs/i,并通过fs/i计算各自的标示含量。以外标法计算4 种成分的标示含量,并对比2 种方法所测得结果的差异。结果见表5。外标法与一测多评法所测得结果的RSD均小于5.0%,差异无显著性,表明一测多评法可用于混合核苷片中多种成分含量的测定。
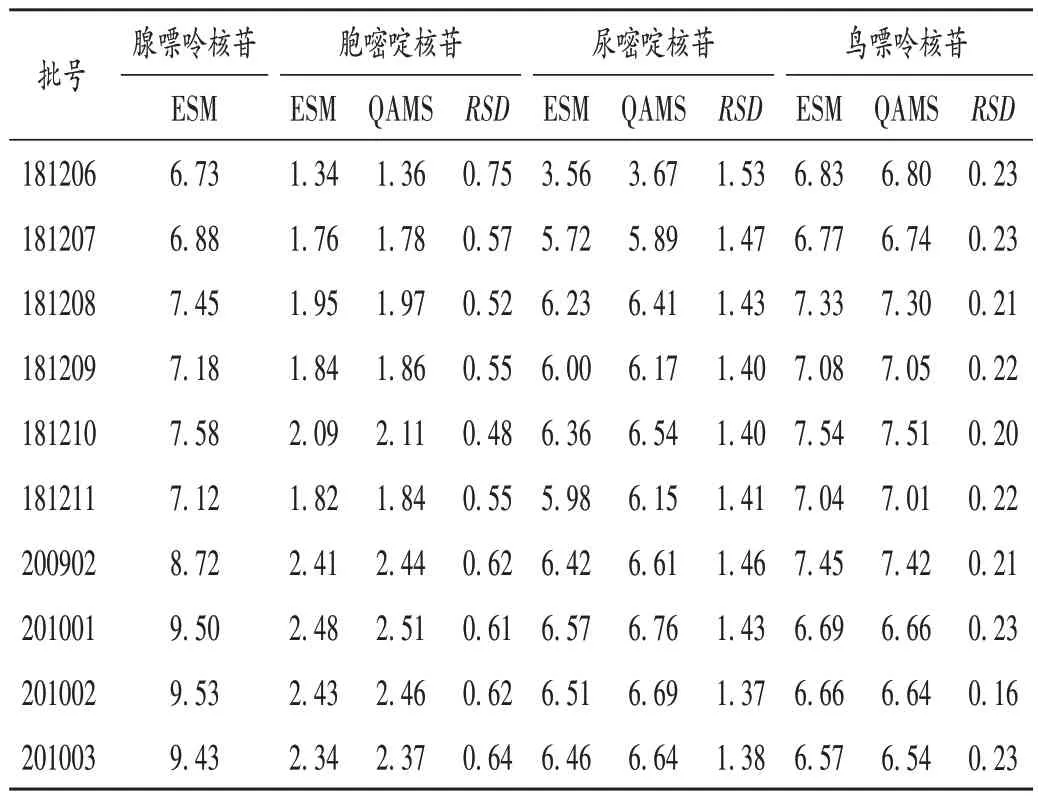
表5 外标法与一测多评法测定结果比较(%)Tab.5 Comparison of content determination of four components in Mixed Nucleoside Tablets by the ESM and the QAMS(%)
3 讨论
混合核苷片中主要有效成分为胞嘧啶核苷、尿嘧啶核苷、鸟嘌呤核苷、腺嘌呤核苷等,其中腺嘌呤核苷对照品易得,性质较稳定。故选择腺嘌呤核苷为内参物。混合核苷片中鸟嘌呤核苷、腺嘌呤核苷、尿嘧啶核苷、胞嘧啶核苷均易溶于水。水作提取溶剂具有干扰小、不污染环境等优点[16]。故选择水提法。本研究中,供试品溶液于200~400 nm 波长范围进行紫外扫描,于257.8 nm 波长处有最大紫外吸收,且空白辅料溶液在此范围内无紫外吸收。参考现行国家标准中含量测定方法,选择260 nm作为最终吸收波长。
综上所述,一测多评法可用于测定混合核苷片中多种成分的含量,可为其质量标准的研究提供参考。
猜你喜欢
临床肝胆病杂志(2022年6期)2022-11-25
广州化工(2022年18期)2022-10-23
河南农业·综合版(2022年2期)2022-03-18
河南农业(2022年2期)2022-03-14
河南农业·综合版(2021年7期)2021-08-23
河南农业(2021年7期)2021-07-30
中国应急管理科学(2021年4期)2021-04-13
昆明医科大学学报(2021年2期)2021-03-29
中西医结合肝病杂志(2020年2期)2020-10-27
中学化学(2019年4期)2019-08-06
