
Full-length transcriptome of anadromous Coilia nasus using single molecule real-time (SMRT) sequencing
2022-08-08 10:24JinpengZhangShufangGaoYonghaiShiYinlongYanQigenLiu
Aquaculture and Fisheries 2022年4期
Jinpeng Zhang, Shufang Gao, Yonghai Shi, Yinlong Yan, Qigen Liu,*
aNational Demonstration Center for Experimental Fisheries Science Education, Shanghai Ocean University, Shanghai, 201306, China
bCentre for Research on Environmental Ecology and Fish Nutrition of the Ministry of Agriculture, Shanghai Ocean University, Shanghai, 201306, China
cShanghai Fisheries Research Institute, Shanghai Fisheries Technology Extension Station, Shanghai, 200433, China
Keywords:
Coilia nasus
SMRT-seq
Alternative splicing
SSR
lncRNA
A B S T R A C T
This study was designed to generate the full-length transcriptome of Coilia nasus using single-molecule long read isoform sequencing (SMRT-seq) technology. RNAs of brains, ovaries, and testes of C. nasus conditioned in both seawater and freshwater except for gut which was only from the fish conditioned in freshwater, were isolated. All the RNAs were pooled together equally for SMRT sequencing. One SMRT cell produced 64.18 Gb clean data,including 765,184 circular consensus (CCS) reads and 616,126 full-length non-chimeric (FLNC) reads. Finally,93,793 non-redundant transcripts were obtained after clustered and polished. Structural analysis of the 93,793 non-redundant transcripts predicted 8242 alternative splicing events, a total of 229,825 SSR and 72,037 complete CDS. A total of 8437 lncRNA were identified. 84,617 transcripts were annotated in NR, Swissprot, GO, COG,KOG, eggnog, Pfam and KEGG database totally. This is the first full-length transcriptome of Coilia nasus,including transcripts appeared in the case of seawater, which will facilitate exploration of genetic data and life story of Coilia nasus.
1.Introduction
TheCoilia nasusis anadromous fish, which is classified as genusCoilia, family Engraulidae, order Clupeiformes.C. nasusis an economically and nutritionally high valued fish in China and once became the most expensive fish in the world due to its tender meat and great taste(Jiang et al., 2017; Xu et al., 2016).C. nasusgrow fattening in the sea from mid-October to February of the following year, during which the gonad remain in the second period (Jiang et al., 2012; Xu et al., 2016).Ovaries and testes are going to develop asC. nasusmigrates into fresh water (Li et al., 2007; Xu et al., 2016; Zhou et al., 2015). Unfortunately,due to over fishing, environmental pollution and other anthropogenic activities, the natural resources ofC. nasushave plummeted (Shen et al.,2015). Fundamental researches are thus essential for this high-valued species to be well conserved or managed and sustainably exploited in the future.
Previous studies mainly focused on nutrition, artificial breeding and culture, life story, geographical distribution (Jiang et al., 2012, 2017; Li et al., 2019) and employed the next generation sequencing to study the testes (Zhou et al., 2015), ovaries (Duan et al., 2015; Song et al., 2014;Xu et al., 2016), olfactory Epithelium (Duan et al., 2015; Zhu et al.,2014), liver, brains (Du et al., 2014; Liu et al., 2019; Wang et al., 2019).As well, there were some researches on gene functions (Fang et al., 2017;Zhu et al., 2017). As an anadromous fish, seawater growth period might be essential for the accumulating of nutrition and preparation for the migration and gonad development. However, studies about seawater growth period ofC. nasusare limited. So are the genetic data ofC. nasusat this stage. Although there are many advantages of next-generation sequencing, such as low price, high throughput and quantifiability,NGS technology is less effective in assembling the full-length transcripts using the short sequencing reads without a reference genome, which might lead to incorrect annotations in some cases (Au et al., 2013;Steijger et al., 2013). Low-quality transcripts obtained through Illumina RNA sequencing limit the scope of analysis of alternative splicing variants and corrected annotation (Wang et al., 2019). In contrast,single-molecule long read isoform sequencing (SMRT-seq) technology can help us gain long-read or full-length transcriptome without assembly and study the structure of mRNAs, which increase genes discovery,alternative splicing and alternative polyadenylation detection and long non-coding RNAs prediction, etc (Sharon et al., 2013; Tilgner et al.,2015; Wang et al., 2019). Fortunately, the genome has been released,lately (Xu et al., 2020). However, full-length transcriptome are still essential to discover new transcripts, assist genome annotation, and identify alternative splicing and gene fusion.
Some studies have proved that internal state of fish can affect their behavior, besides migration (Lennox et al., 2019). And from the life-history ofC. nasuswe mentioned above, we believe that seawater may affect the development ofC. nasus, and the regulation of gonad development might be the internal cause of its migration behavior. And brain-gonad axis is the key regulatory factor of gonad development. So we studied the cDNA library of pooled brains, testes and ovaries from both freshwater and seawater breedingC. nasus, besides gut from freshwaterC. nasusin Shanghai Fisheries Research Institute by making use of SMRT-seq. This is the firstC. nasustranscriptome constructed with PacBio, which include transcripts appeared in sea situation. This study might be used as supplementary resource of the genome for investigation on the structure of mRNAs and the discovery of new genes ofC. nasus.
2.Materials and methods
2.1.Experimental fish and sample collection
TheC. nasuswere offered by aquafarm of Shanghai Fisheries Research Institute in Fengxian, Shanghai (30°51′42.66′′N; 121°43′4.72′′E). From November 2018 to mid-February 2019,C. nasuswere cultured in seawater and fresh water respectively. The seawater was pumped from the estuary area, with a salinity of 9‰, and increased to 25‰ using solar salt. The fish were anesthetized with a dose of 30 mg/L of eugenol(Maclin, China). Brains, ovaries and testes were sampled from both freshwater- and seawater-treated fish, besides freshwater gut. Tissues were promptly placed into liquid nitrogen and stored at -80°C before RNA extraction.
2.2.Total RNA extraction, library construction and SMRT sequencing
Total RNAs were isolated from both freshwater and seawater brains,ovaries and testes ofC. nasus, besides freshwater gut, respectively, by using TRIzol®Reagent (Vazyme, China) according to the directions.Equal quantities of total RNAs from the seven RNA samples were pooled for PacBio Isoform Sequencing (Iso-Seq) library construction. Total RNA was reverse transcribed into cDNA by the SMARTer PCR cDNA Synthesis Kit (Clontech, USA). Subsequently, PCR amplification was carried out by PrimeSTAR GXL DNA polymerase (Takara, Japan) through taking reverse transcription cDNA as a template. The PCR products were purified with 1X and 0.4X AMPurePB magnetic beads (PacBio, USA)severally. The concentrations and sizes of the purified products were detected. The 1X and 0.4X purified products were mixed based on size by the same molar mass. Then, Iso-Seq library was constructed by SMRTbell Template Prep Kit (PacBio, USA). The library was purified with AMpure PB magnetic beads (PacBio, USA), concentration and size were detected. Finally, qualified and adequate library ligated with primer and polymerase using PacBio Binding kit (PacBio, USA). The final cDNA library was sequenced on PacBio Sequel (Biomarker Technologies, Beijing, China).
2.3.Pacific biosciences long read processing
Subreads were gained by filtering the polymerase reads with fragment length<50 bp and a quality of<0.90, and taking the left sequences’ adaptors away. Discarded subreads that were<50 bp in length,and the remaining subreads are clean data. Circular consensus sequences (CCS) were extracted from the clean data with full passes of ≥1 and a quality of>0.90. The CCS reads were divided into full-length sequences (including 5’/3′primers and polyA tails) and non-fulllength sequences by detecting whether the CCS reads contained the correct 5’/3′primers and polyA tails. Full length non-chimeric reads(FLNC) were determined according to the 5’/3′primers and poly A tails in the full-length sequences. Consensus isoforms in FLNC were obtained and polished by using SMRTLink software. High quality full-length transcripts were acquired by the criteria of post-correction accuracy>99%.
2.4.Remove redundant
CD-HIT (Weizhong & Adam, 2006) software was used to remove redundant sequences from high quality full-length transcripts. The left non-redundant transcripts constitute the full-length transcriptome. The integrity of the transcriptome was evaluated by BUSCO (Simão et al.,2015).
2.5.Structure analysis
2.5.1.Simple sequence repeat (SSR) detection
We used MISA (http://pgrc.ipk-gatersleben.de/misa/) to identify 7 types of SSR by analyzing the transcripts that are>500 bp. Seven types of SSR are mono-nucleotides, di-nucleotides, tri-nucleotides, tetra-nucleotides, penta-nucleotides, hexa-nucleotides and compound SSR.
2.5.2.CDS detection
TransDecoder software (https://github.com/TransDecoder/Trans Decoder/releases) was used to predict the coding sequences of transcripts. It uses the following criteria: firstly, the shortest open reading frame (ORF) in a transcript sequence is found. Secondly, using the GeneID software, a log-likelihood score similar to what is computed is>0. Thirdly, when the ORF is scored in the first reading box, the above coding score is the largest compared with the scores in other reading boxes. Fourthly, if one candidate ORF is completely covered by the other one, the longer one determined. However, a single transcript may report a plurality of open reading frames (considering operon, chimeras, etc.).Finally, the predicted peptide matches the Pfam domain higher than the noise cutoff fraction.
2.5.3.Alternative splice (AS) prediction
Transcripts were in pairs compared by BLAST software (Altschul et al., 1998). Generally, it can be considered as a candidate AS event, if the compare result meets the following conditions simultaneously (Liu et al., 2017): firstly, both transcripts are longer than 1000 bp, and there are two HSPs (High-scoring Segment Pairs) in the two sequences. Secondly, AS gap is longer than 100 bp and be away from at least 100 bp from the 3 ’/5′end. Thirdly, all alternative transcripts can be allowed to have an overlap of 5 bp.
2.6.Prediction of lncRNA and targeted transcripts of lncR NA
Coding Potential Calculator (CPC) (Kong et al., 2007),Coding-Non-Coding Index (CNCI), Coding Potential Assessment Tool(CPAT) (Wang et al., 2013), and pfam protein structure domain analysis are the most widely used four methods that predict lncRNA. Here, we integrated the four methods to predict lncRNA of transcripts. In addition, LncTar (Li et al., 2014) was used to predict targeted transcripts of lncRNA.
2.7.Functional annotation of transcripts
BLAST software (version 2.2.26) (Simão et al., 2015) was used to gain the annotation information of non-redundant transcripts with mapping NR, Swissprot, GO, COG, KOG, eggnog, Pfam and KEGG database.
3.Results
3.1.The statistics of SMRT sequencing data
One SMRT cell generated 64.18 Gb clean data. With full passes of ≥1 and a quality of>0.90, a total of 765,184 CCS sequences were obtained(Table 1). Subsequently, 616,126 FLNC sequences were determined(Table 2). 161,195 polished high-quality isoforms were gained by using SMRTLink software (Table 3). Finally, 93,793 non-redundant transcripts were obtained by CD-HIT software. We used BUSCO pipeline to check the completeness of non-redundant transcripts. The result indicated that 84.6% of the non-redundant transcripts (4584 genes) were complete. In 4584 genes, 28.8% (1319 genes) were complete single-copy BUSCOs,55.8% (2556 genes) were complete duplicated BUSCOs, 4.1% (186 genes) were fragmented BUSCOs, and 11.3% (523 genes) were missing BUSCOs. The generated PacBio SMRT reads have been submitted to the NCBI Sequence Read Archive (SRA) (accession number SRR10512642).

Table 1Statistics of CCS data.
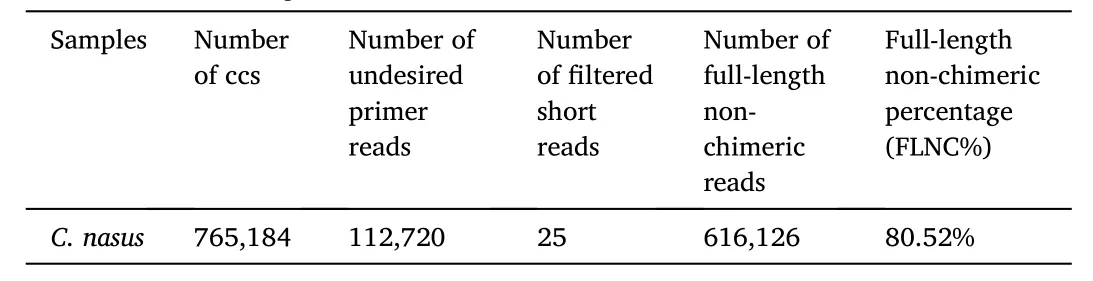
Table 2Statistics of full-length reads.
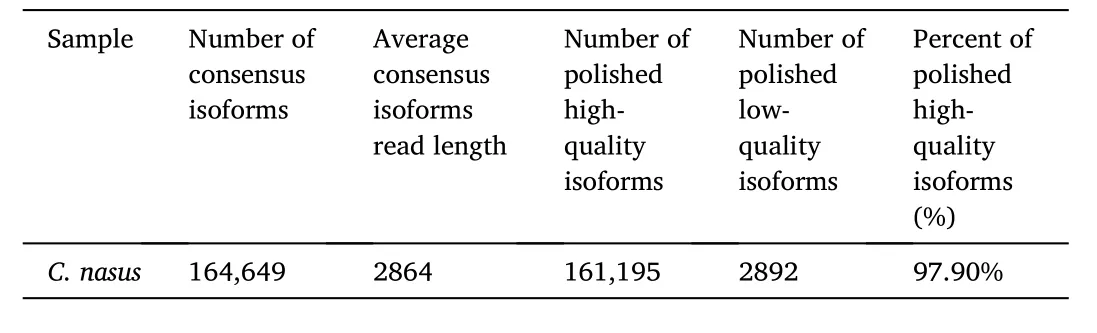
Table 3Statistics of consensus isoforms.
3.2.Structure analysis
3.2.1.SSR detection
A total of 92,759 sequences (285, 799, 680 bp) were used for SSR analysis. 229,825 SSR were detected, including 63,959 SSR containing sequences. 44,523 sequences contain more than one SSR, and 76,860 SSR appear in compound formation. Additionally, the numbers of mononucleotides, di-nucleotides, tri-nucleotides, tetra-nucleotides, pentanucleotides, and hexa-nucleotides were 65,275, 133,810, 22,850,6,023, 1340 and 527, respectively.
3.2.2.CDS detection
82,802 ORFs were identified, including 72,037 complete ORFs by using TransDecoder software. The length distribution of detected CDS is exhibited in Fig. 1.
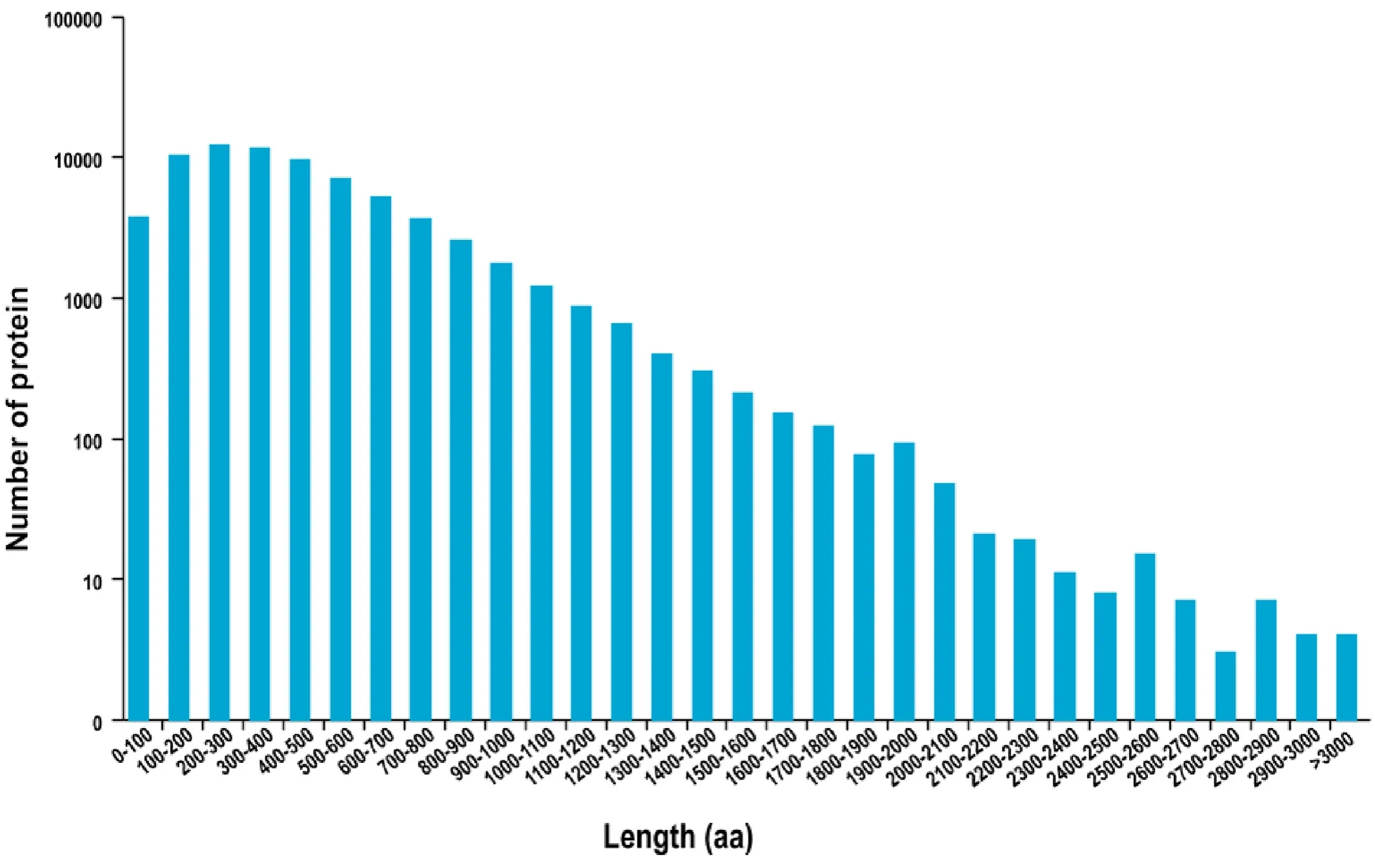
Fig. 1.The coding protein length distribution of the predicted CDS.
3.2.3.AS prediction
Pre-mRNA formed from template DNA can have multiple splicing.Different exons are selected to produce different mRNAs, which can code different proteins, which result in the diversity of biological traits.This procedure is called alternative splicing. We employed the nonredundant transcripts to foretell Alternative splicing candidate events.With the criteria mentioned above in methods, we identified 8242 AS events.
3.3.Prediction of lncRNA and targeted transcripts of lncRNA
Combined CPC, CNCI, CPAT and pfam protein structure domain four methods, 8437 lncRNA transcripts were predicted totally (Fig. 2). Using LncTar tool, 3771 lncRNA were predicted to have target transcripts(Supplemental Tab 4).

Fig. 2.Venn diagram of the number of lncRNAs identified by CNCI, CPC, Pfam and CPAT.
3.4.Functional annotation of transcripts
In all, 84,617 transcripts were annotated in the eight databases,26,422 in the COG database, 56,022 in GO, 56,530 in KEGG, 63,968 in KOG, 72,637 in Pfam, 57,414 in SwissProt, 82,843 in eggnog, and 84,188 in NR. Homologous species ofC. nasuswere identified through sequence alignment by NR. About 23.11% of sequences were aligned toAstyanax mexicanus, which is the most matched species, followed byDanio rerioandEsox luclus(Fig. 3). The GO classification statistics of non-redundant transcripts are shown in Fig. 4. Top three GO terms are cell part, cell and cellular process.
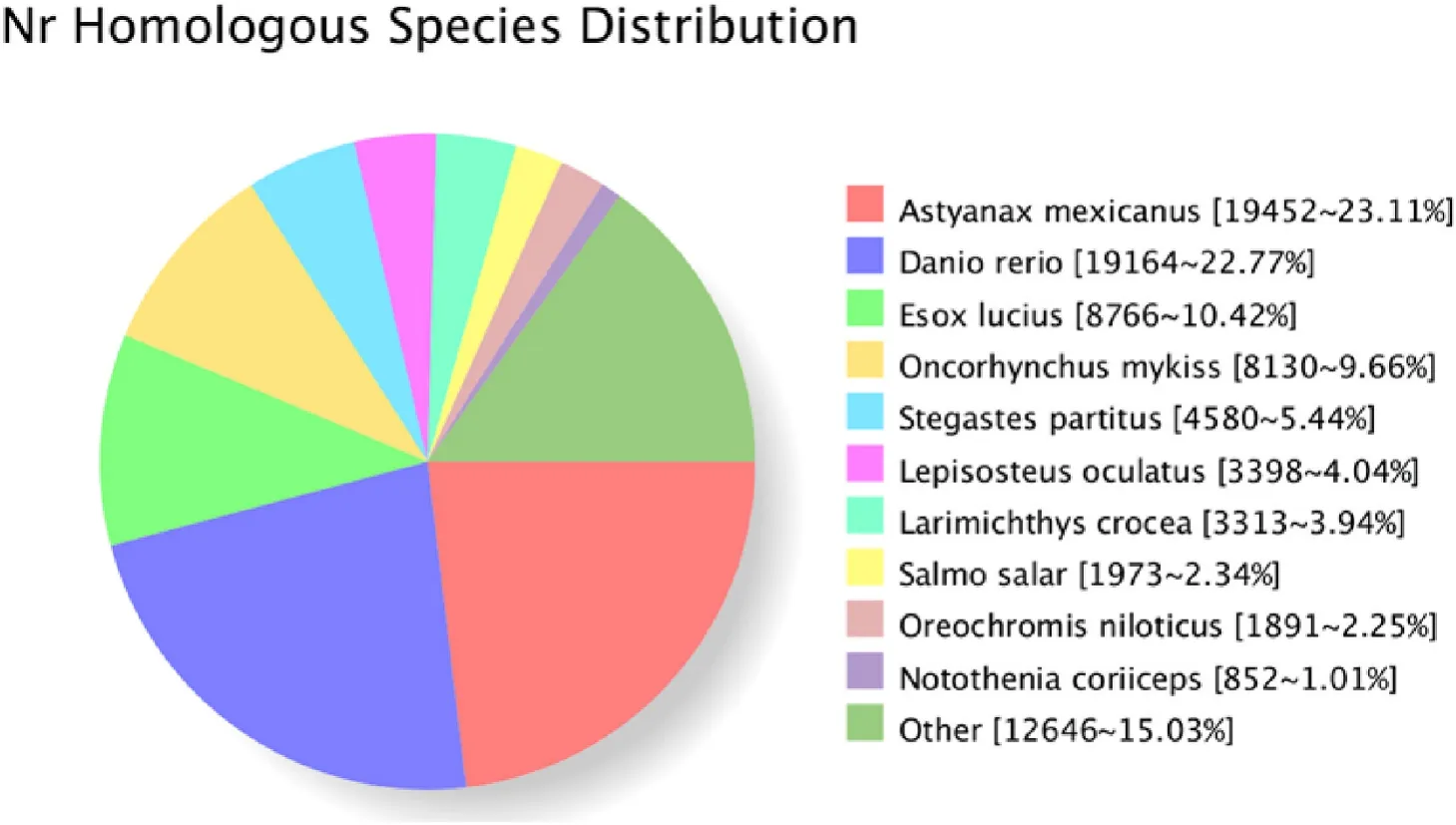
Fig. 3.The NR homologous species distribution of the consensus isoforms.
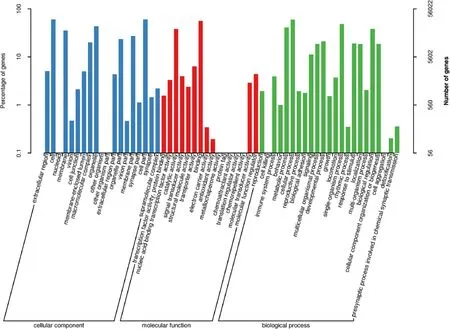
Fig. 4.The GO function annotation of the consensus isoforms. The abscissa is the GO classification, the left of the ordinate is the percentage of the number of genes,and the right is the number of genes.
4.Discussion
Transcriptome research is one of the indispensable tools for understanding life processes. However, without a reference genome, rna-seq technology based on NGS cannot accurately obtain or assemble complete transcripts, and recognize the isoform, homologous genes and superfamily genes, which make it dif ficult to understand the deeper meaning of life activities. Full-length transcriptome sequencing based on PacBio SMRT single-molecule real-time sequencing technology does not interrupt RNA fragments, and direct reverse transcription to obtain fulllength cDNA. The platform’s ultra-long reading (median 10 kb) contains complete transcripts’ information without assembly (Gordon et al.,2015; Thomas et al., 2014), which can help us obtain the initial transcripts of life activities.C. nasusis of great value because of its tender meat and delicious taste. Furthermore, due to its diverse ecological types, it has important scientific value for studying the adaptive evolution of fish migration. However, the amount ofC. nasushas decreased sharply, and artificial breeding technology is immature yet, which limited the availability ofC. nasus. Rare resources and the strong reaction to little stimulation ofC. nasusmake it dif ficult to obtain high quality RNA. In these circumstances, building full-length transcriptome library is essential. Even though the genome has recently been released(Xu et al., 2020), the full-length transcriptome is indispensable for more accurate at the prediction of transcripts, alternative splicing, fusion genes, gene families, and non-coding RNAs than the genome.
In this study, PacBio platform was used to sequence and analyze the full-length transcriptome of pooled RNA of brains, testes, ovaries and gut. One SMRT cell produced 64.18 Gb clean data. After processing, we obtained 93,793 non-redundant transcripts. By structure analysis of the non-redundant transcripts, we identified 8242 alternative splices,229,825 SSR and 72,037 complete CDS. 8437 lncRNA were predicted.84,617 transcripts were annotated. The density distributions of different types of SSR are shown in Fig. 5. The number of mono-nucleotide (p1)and di-nucleotide (p2) is the largest, which are 168 and 154 per Mb respectively and they are significantly higher than other types of SSR,followed by compound SSR, 135 per Mb. Tri-nucleotide, tetra-nucleotide, penta-nucleotide and hexa-nucleotide are 59, 10.6, 3, 1 per Mb separately. The predicted SSR are obviously more than data based on next-generation transcriptome sequencing (Shen et al., 2015; Zhu et al.,2014). These data can lay the foundation for population genetic studies,genetic linkage map construction, and breeding studies ofC. nasus(Grover & Sharma, 2016).

Fig. 5.Statistics of density distribution of seven SSR types. C, compound SSR; p1, mono-nucleotide; p2, di-nucleotide; p3, tri-nucleotide; p4, tetra-nucleotide; p5,penta-nucleotide; p6, hexa-nucleotide.
In eukaryotes, the coding RNA account for a small proportion.LnRNA, as one of the most studied non-coding RNAs, is rarely studied in teleost (Al-Tobasei et al., 2016; Pauli et al., 2012; Wang et al., 2016). A large amount of evidence has proved the important roles of lncRNA in transcriptional, post-transcriptional levels and cellular or physiological processes at epigenetic (Kretz et al., 2013; Nakagawa & Kageyama,2014). This study not only identified a large number of lncRNA, but also predicted targeted transcripts of lncRNA, which provided significant data for transcriptional, post-transcriptional and epigenetic research ofC. nasus.
In organisms, gene products interact with each other to carry out biological functions. Annotation analysis of pathway for gene expression helps to further interpret the function of genes. In this study, 56,530 transcripts annotated by the KEGG are attributed to 297 pathways,including fatty acid biosynthesis and metabolism, steroid biosynthesis,steroid hormone biosynthesis etc. These pathways are extremely important for the further study ofC. nasus.
5.Conclusion
In this study, we built the full-length transcriptome ofC. nasusfor the first time, including transcripts that appeared in seawater, which enhanced our understanding of the gene structure and can be used as supplementary data of genome for fundamental research ofC. nasus.
CRediT authorship contribution statement
Jinpeng Zhang: Investigation, Data curation, Writing - original draft. Shufang Gao: Investigation, Data curation. Yonghai Shi: Project administration, Resources, Supervision. Yinlong Yan: Resources,Funding acquisition. Qigen Liu: Project administration, Writing - review & editing, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to infuence the work reported in this paper.
Acknowledgments
This work was supported by the Project of Centre for Research on Environmental Ecology and Fish Nutrition (CREEFN) of the Ministry of Agriculture and Rural Affairs, Shanghai Ocean University (A1-3201-19-300204) and the Project of Science and Technology Commission Shanghai Municipality (17391900300).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.aaf.2020.08.006.
 Aquaculture and Fisheries2022年4期
Aquaculture and Fisheries2022年4期
- Aquaculture and Fisheries的其它文章
- A review on fishing gear in China: Selectivity and application
- atg7 and beclin1 are essential for energy metabolism and survival during the larval-to-juvenile transition stage of zebra fish
- Association between single nucleotide polymorphisms of nLvALF1 and PEN2-1 genes and resistance to Vibrio parahaemolyticus in the Pacific white shrimp Litopenaeus vannamei
- Comparative analysis of the morphology, karyotypes and biochemical composition of muscle in Siniperca chuatsi, Siniperca scherzeri and the F1 hybrid (S. chuatsi ♀ × S. scherzeri ♂)
- Effects of salinity and alkalinity on fatty acids, free amino acids and related substance anabolic metabolism of Nile tilapia
- Differences in postembryonic dorsal fin development resulted in phenotypic divergence in two gold fish strains, Red Cap Oranda and Ranchu