
TRPV4在心肌细胞缺氧损伤中的作用及机制研究*
2022-08-05 03:03何玥颖谢高倩苗路伟陈克明
中国病理生理杂志 2022年7期
陈 卓, 秦 昆, 何玥颖, 魏 朋, 谢高倩, 苗路伟, 陈克明△
(1解放军联勤保障部队第940医院基础医学实验室,甘肃 兰州 730050;2兰州理工大学生命科学与工程学院,甘肃 兰州 730050)
瞬时受体电位阳离子通道V 亚家族成员4(transient receptor potential cation channel subfamily V member 4,TRPV4)是瞬时受体电位(transient receptor potential,TRP)通道蛋白家族中类香草素亚家族的成员之一[1]。TRP 家族有近 30 个亚型,TRPV4 作为TRP 家族一种非选择性的阳离子通道而在多种细胞内的生理过程中发挥作用[2]。现已有研究证明,TRPV4在渗透压变化、热、细胞肿胀和化学刺激等过程被激活而发挥作用[3-6]。
高原缺氧后心肌细胞的氧化应激系统被激活,导致细胞产生大量的活性氧(reactive oxygen species,ROS),最终引起急性心肌梗死[7]。目前的研究认为Ca2+与ROS 的产生有关。已经有研究证明在心脏缺血再灌注损伤中TRPV4 蛋白水平大量升高,并引起心脏收缩增强[8]。此外,Ca2+过度升高与 ROS 大量产生在急性呼吸窘迫综合征中也起着至关重要的作用[9]。还有研究证明,TRPV4 诱导神经元和星形胶质细胞损伤是通过Ca2+超载和氧化应激[10]。
本实验将通过使用TRPV4 抑制剂HC-067047,探究缺氧后心肌细胞中TRPV4 的作用机制,及其是如何调控氧化应激。
材料和方法
1 仪器
MIC-101 型低氧处理盒(Billups-Rothenberg);IX71 型倒置荧光显微镜(Olympus);EPOCH 型酶标仪(BioTek);Labofuge 400R 型离心机(Heraeus);165-8033 型电泳仪(Bio-Rad);高分辨率活细胞成像(Cytiva);RT-PCR仪(ViiATM7);细胞培养箱(Themo)
2 细胞来源
H9C2细胞购于ATCC。
3 试剂
胎牛血清购自 Biological Industries;DMEM 培养基和胰蛋白酶均购于Gibco;CCK-8 试剂购于北京酷来博科技有限公司;ROS 试剂盒、过氧化氢酶(catalase,CAT)试剂盒、超氧化物歧化酶(superoxide dismutase,SOD)试剂盒和丙二醛(malondialdehyde,MDA)试剂盒均购于南京建成生物工程研究所;线粒体膜电位试剂盒(JC-1)购于碧云天公司;RIPA 高效裂解液、非变性裂解液、ECL 发光液、BCA 蛋白浓度测定试剂盒、SDS-PAGE 凝胶制备试剂盒和Fluo-4 AM试剂盒均购于Solarbio;兔抗TRPV4抗体、兔抗βactin 抗体及山羊抗兔IgG Ⅱ抗均购于Abcam;SYBR Green Premix Pro Taq HS qPCR Kit 和Evo M-MLV RT Kit with gRNA Clean for qPCR 均购于 Accurate Biology。
4 实验方法
4.1 细胞培养 使用含10%胎牛血清的DMEM 培养基,在含5% CO2的37℃饱和湿度培养箱内,每3 d更换培养基,细胞密度约80%时传代培养。
4.2 细胞的缺氧处理 将低氧处理盒置于培养箱内,取H9C2 细胞换成含1%的胎牛血清的DMEM 培养基后放入低氧盒,打开气体阀门向盒内通入95%N2和5% CO2,检测氧气浓度到达1%时,关闭阀门,密封处理盒,并开始计算缺氧时间。倒序0 h、12 h、24 h 和36 h 分别放入H9C2 细胞,待缺氧时间完成后一起取出细胞进行下一步检测。
4.3 细胞HC-067047 处理 待细胞长至80%,缺氧处理前 2 h 加入 1 μmol/L 的 HC-067047,对照组加入同等含量的DMSO。
4.4 CAT、SOD 和 LDH 活性及 MDA 含量的测定将H9C2细胞种植于60 mm 培养皿中,待长至80%进行缺氧处理。缺氧处理后H9C2 细胞使用非变性裂解液裂解后,15 000 r/min 离心25 min,吸取上清,使用BCA 试剂盒检测蛋白浓度,再根据CAT、SOD、MDA 和 LDH 试剂盒说明书检测 CAT、SOD 和 LDH 活性及MDA 含量,并使用酶标仪在不同的波长下进行数据统计分析。
4.5 CCK-8 法检测细胞活力 首先按照每孔3 000个细胞种植于96 孔板中,缺氧处理后加入含有10 μL CCK-8 溶液,孵育2 h 后使用酶标仪检测,按照说明书计算细胞相对活力。
4.6 细胞内ROS 检测 将H9C2 细胞种植于60 mm培养皿中,待长至80%进行缺氧处理。缺氧处理后弃去培养基,PBS 洗 2 遍后加入2 mL DCFH-DA 工作液。培养箱中孵育1 h。取出后PBS洗3遍。荧光显微镜观察荧光变化,荧光酶标仪最佳激发波长485 nm,最佳发射波长525 nm 检测ROS 荧光强度,并根据说明书计算ROS水平。
4.7 Western blot 检测TRPV4 蛋白水平 将H9C2细胞种植于60 mm 培养皿中,待长至80%进行缺氧处理并提前2 h 加入HC-067047。缺氧处理后弃去培养基,PBS 洗2 遍,RIPA 裂解液裂解细胞后提取蛋白,15 000 r/min 离心 25 min 后取上清,BCA 试剂盒测定蛋白浓度,加入4×上样缓冲液沸水煮10 min 使其变性。SDS-PAGE 凝胶制备试剂盒配制10%分离胶和5%的浓缩胶分离蛋白,采用湿转法将蛋白转移至PVDF 膜上。使用5%的脱脂奶粉使膜封闭2 h,TRPV4(1∶1 000)和 β-actin(1∶10 000)Ⅰ抗孵育过夜。孵育结束后使用TBST洗膜每遍10 min,共4次。接着加入Ⅱ抗(1∶1 000)室温孵育2 h,TBST洗膜4次后曝光,加ECL发光液扫描条带并进行分析。
4.8 RT-qPCR 检测 TRPV4 的 mRNA 表达量 将H9C2 细胞种植于60 mm 培养皿中,待长至80%进行缺氧处理提前2 h 加入HC-067047。缺氧处理后弃去培养基,每皿加入1 mL RNAiso Plus 提取细胞内RNA。再使用有机溶剂法提取细胞内总RNA,接着使用Evo M-MLV RT Kit with gRNA Clean for qPCR 将其反转录后,使用SYBR Green Premix Pro Taq HS qPCR Kit进行RT-qPCR操作。并使用β-actin作为内参对结果进行校正,用2-ΔΔCt表示各基因的相对表达水平。引物序物见表1。

表1 RT-qPCR引物序列Table 1. Primer sequence for RT-qPCR
4.9 使用Fluo-4 AM检测Ca2+水平 Fluo-4 AM首先用Pluronic F127 溶液等量溶解。用HBSS 稀释Fluo-4 AM 溶液,制备4 μmol/L 工作液。将工作液加入H9C2细胞中,37 ℃孵育20 min,然后加入5倍体积含1%胎牛血清的HBSS,再培养40 min。用HEPES 洗涤后,用荧光酶标仪检测钙离子浓度。最佳激发波长和发射波长分别为506和526 nm。
4.10 线粒体膜电位试剂盒(JC-1)检测线粒体膜电位 线粒体膜电位高时产生红色荧光,线粒体膜电位低时产生绿色荧光。用红绿荧光的相对比值测定线粒体膜电位。将H9C2 细胞种植于60 mm 培养皿中,待长至80%进行缺氧处理提前2 h 加入HC-067047。缺氧处理后弃去培养基,用PBS 洗涤待染色细胞。加入JC-1 染色液,37 ℃孵育20 min。孵育后用JC-1染色缓冲液洗涤,加入适量DMEM 培养基,用荧光显微镜和荧光素微孔板观察。JC-1 单体的最佳激发光和发射光分别为490 和530 nm;JC-1 聚集体的最佳激发光和发射光分别为525和590 nm。
5 统计学处理
数据以均数±标准差(mean±SD)表示。采用SPSS 20.0 进行统计分析。数据的统计差异采用单因素方差分析(one-way AVNOVA)。以P<0.05 为差异有统计学意义。
结 果
1 H9C2细胞在缺氧环境发生氧化性损伤
与常氧组相比,随着缺氧时间的延长,H9C2 细胞活力发生明显下降(P<0.01),见图1A。此外,LDH 溢出率和MDA 含量也随着缺氧时间增多,并在24 h 达到峰值(P<0.01),见图1B、C。同时检测了ROS,发现绿色荧光随着缺氧时间明显变强,见图1D。并且H9C2 细胞中细胞内抗氧化酶CAT 和SOD的活性显著提升(P<0.01),见图1E、F。在缺氧24 h时H9C2 的氧化应激被激活得最为明显,因此选择24 h为后续实验时点。
2 H9C2细胞在缺氧环境发生钙超载和线粒体损伤
已有研究证明钙离子与氧化应激有着密不可分的关系。我们发现H9C2 细胞缺氧24 h 后绿色荧光明显增多,钙离子含量明显增高,见图2A。JC-1 检测线粒体膜电位,缺氧24 h后绿色荧光明显增多,即线粒体膜电位明显下降,见图2B。以上结果说明H9C2在缺氧环境发生钙超载和线粒体损伤。
3 钙超载是由TRPV4高表达引起
TRPV4 是非选择性的钙离子通道。我们猜想H9C2 细胞中的钙超载可能与TRPV4 有关。实验发现缺氧后TRPV4无论是蛋白水平还是mRNA水平均显著上调(P<0.01),见图3A、B。并且使用TRPV4激活剂GSK1016790A 检测钙离子水平变化,结果显示加入激活剂后的H9C2 细胞缺氧处理后比常氧情况下Ca2+有较大涨幅,见图3C。
4 抑制TRPV4 对H9C2 细胞缺氧后氧化应激系统的影响
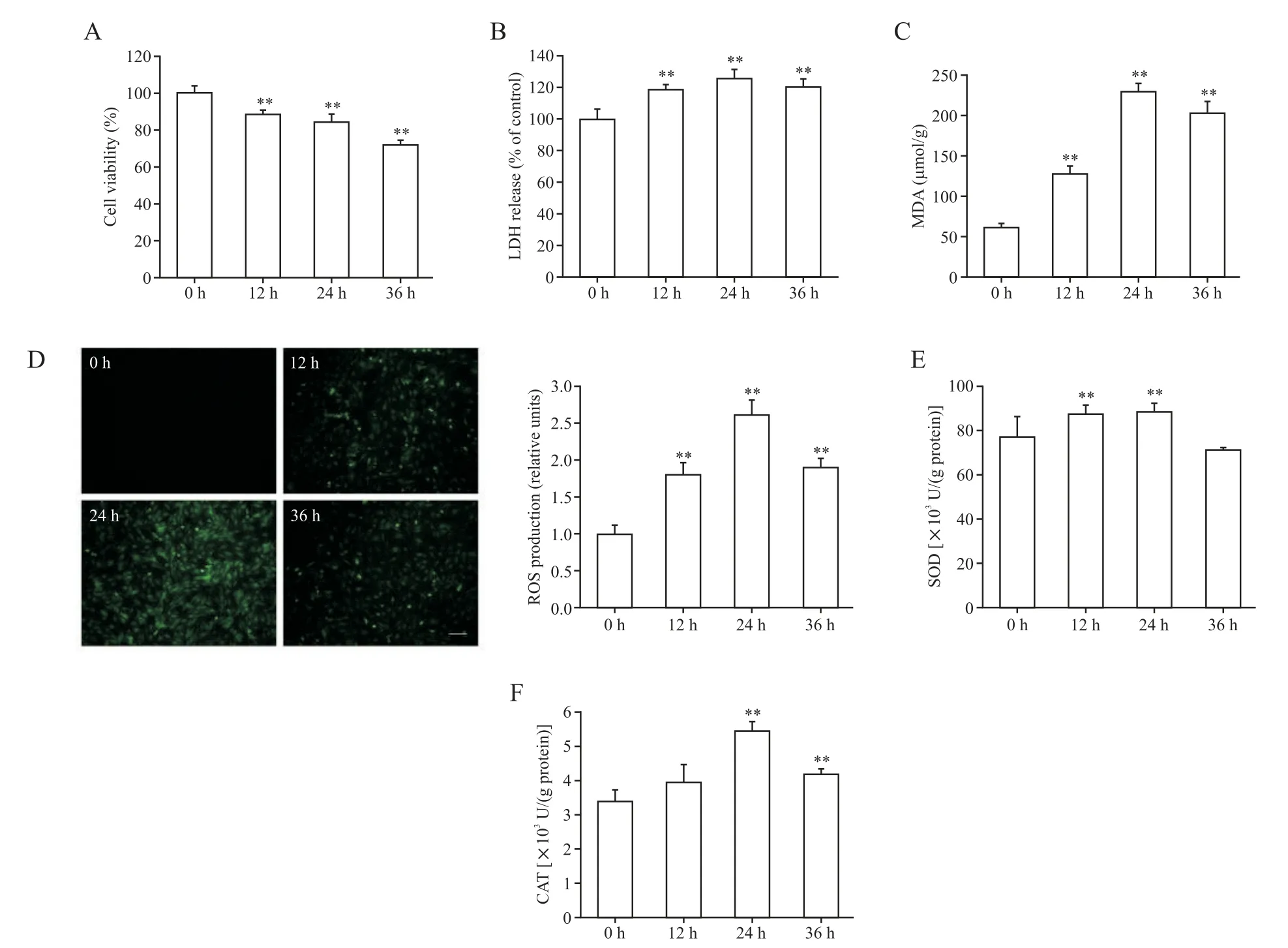
Figure 1. Prolonged hypoxia significantly increased oxidative damage of H9C2 cells. A:cell viability;B:LDH release rate;C:the MDA content;D:ROS fluorescence intensity(scale bar=100 μm);E:SOD activity;F:CAT activity. Mean±SD. n=3.*P<0.05,**P<0.01 vs 0 h group.图1 缺氧不同时点对H9C2氧化损伤影响

Figure 2. Calcium overload and mitochondrial damage occurred in H9C2 cells after hypoxia(scale bar=50 μm). A:green fluorescence represents calcium ion;B:red fluorescence represents JC-1 aggregates,and green fluorescence represents JC-1 monomer.图2 缺氧24 h后观察H9C2细胞内Ca2+和线粒体膜电位的变化
既然缺氧后TRPV4在蛋白和mRNA 水平均显著上调,为了探究TRPV4 在H9C2 缺氧后起的作用,我们使用了TRPV4 抑制剂HC-067047 抑制TRPV4,并从mRNA和蛋白水平证明使用抑制剂后TRPV4被抑制,见图4A、B。与缺氧24 h 相比,加入抑制剂后无论是荧光照片还是量化结果ROS 水平均显著下降,见图4C;同时抗氧化酶CAT 和SOD 活性也均出现下降趋势,见图4D、E。此外,缺氧后细胞相对活力下降到80%,加入抑制剂后细胞活力显著上升,见图4F。上述结果表明,TRPV4在H9C2细胞缺氧时被激活并调控氧化应激系统。
5 TRPV4 通过调节钙离子起作用并会影响线粒体膜电位
为了进一步证明TRPV4调控氧化应激是否通过Ca2+,我们检测了使用TRPV4抑制剂HC-067047后细胞内Ca2+和线粒体膜电位的变化。结果显示,缺氧后钙离子含量明显增高,抑制TRPV4 后钙离子含量也随之下降,见图5A。同时缺氧后线粒体膜电位明显下降,但是抑制TRPV4后线粒体膜电位出现回升,见图5B。以上实验结果说明缺氧状态下,TRPV4 会导致H9C2细胞钙离子升高并影响线粒体膜电位。

Figure 3. Activation of TRPV4 after hypoxia. A:the protein level of TRPV4;B:the mRNA level of TRPV4;C:calcium ion level after adding TRPV4 activator GSK1016790A(GSK). Mean±SD. n=3.**P<0.01 vs 0 h group.图3 缺氧前后TRPV4蛋白和mRNA表达水平的变化及Ca2+水平

Figure 4. The addition of TRPV4 inhibitor HC-067047 significantly inhibited the damage of H9C2 cells after hypoxia. A:the protein level of TRPV4;B:the mRNA level of TRPV4;C:ROS fluorescence intensity(scale bar=100 μm);D:SOD activity;E:CAT activity;F:cell viability. Mean±SD. n=3.*P<0.05,**P<0.01 vs 0 h group;#P<0.05,##P<0.01 vs NC group.图4 加入TRPV4抑制剂对H9C2氧化应激系统的影响
讨 论
氧气是心肌细胞活动不可或缺的物质,一旦心肌缺氧,会直接导致心肌细胞有氧活动减弱,代谢活动减弱,使得心脏必须的能量供给不足,直接导致心功能下降[11]。同时代谢废物不能及时清除也进一步对心肌细胞造成伤害[12]。心肌细胞在缺氧的环境下会启动氧化应激,增强抗氧化酶SOD 和CAT 等的活性。SOD 是一种广泛存在于生物体内的一种抗氧化酶,可催化ROS生成水和过氧化氢;几乎所有的生物体内都含有CAT,它能将过氧化氢催化生成水和氧气,SOD 和 CAT 共同维持氧化与抗氧化平衡[13]。但是心肌细胞如何感知缺氧尚未解释清楚。

Figure 5. After the addition of TRPV4 inhibitor HC-067047,intracellular calcium ion level decreased and mitochondrial membrane potential returned to normal. A:calcium ion level;B:mitochondrial membrane potential. The scale bar=50 μm. Mean±SD. n=3.**P<0.01 vs 0 h group;##P<0.01 vs NC group.图5 加入TRPV4抑制剂对Ca2+和线粒体的影响
本实验首先建立H9C2细胞缺氧模型,发现随着缺氧时间的延长,细胞活力、LDH 释放率、MDA 含量及ROS 水平等结果证实氧化损伤逐步加重,并在24 h 达到顶峰;同时抗氧化酶CAT 和SOD 的活性随着缺氧时间的延长也均显著提高,并在24 h达到顶峰。值得注意的是,到36 h会出现减弱,这可能是由于氧化与抗氧化是一个动态平衡的过程,24 h 为整个缺氧过程中的一个重要拐点。因此后续我们选择24 h为缺氧时点。
近年来的研究发现,Ca2+内流会促进ROS 生成,同时线粒体作为细胞内产生最多ROS的细胞器也被认为与氧化损伤有关[14]。本实验结果证明,H9C2 细胞缺氧24 h 后,细胞内Ca2+含量大量增高,同时线粒体膜电位受损,这说明H9C2细胞的氧化应激很有可能与Ca2+有关。TRPV4 是一个Ca2+高渗透性的阳离子通道。在近年来的研究发现,在许多细胞(如尿路上皮细胞和巨噬细胞等)中激活TRPV4 会导致Ca2+内流,从而促进 ROS 生成[15-16]。我们发现 H9C2 细胞缺氧后TRPV4 的mRNA 和蛋白水平均有所升高,这说明在缺氧过程中TRPV4 被激活。为了探究Ca2+超载是否是由于TRPV4 通道激活导致,我们使用TRPV4 激活剂后Ca2+升高程度明显增强。上述结果说明H9C2 缺氧后会激活TRPV4 并导致Ca2+超载和线粒体受损。
为了进一步探究在H9C2 细胞缺氧后TRPV4 的作用机制,我们使用了TRPV4 特异性抑制剂HC-067047。首先我们验证了加入抑制剂后TRPV4 的mRNA 和蛋白均被抑制。在缺氧环境下,TRPV4 被激活,ROS 含量明显增多,CAT 和 SOD 活性也明显上升,说明细胞受到了氧化损伤,细胞活力降低也证实了这一点。但是当抑制TRPV4 之后,所有的一切都被逆转了,ROS 显著降低,CAT 和 SOD 活性也显著下降,细胞活力也明显上升。这说明在H9C2 细胞中,缺氧会激活TRPV4进而导致氧化应激的产生。
已有研究证明,在血管内皮细胞中TRPV4 激活导致钙离子内流是诱发线粒体ROS增多的重要原因之一[17]。此外,Ca2+超载和过度累积的ROS会导致线粒体膜电位去极化,进而加速细胞死亡[18]。为了进一步确认在H9C2 细胞中TRPV4 的作用机制是否是通过Ca2+和线粒体,我们首先检测了H9C2 细胞中的Ca2+,结果显示缺氧后心肌细胞钙离子含量明显升高,这与之前的结果一致,并且在加入HC-067047 后被显著抑制。接着使用JC-1 检测钙离子膜电位,缺氧24 h后红绿荧光比例下降,说明H9C2细胞线粒体受损,当抑制TRPV4后,线粒体膜电位恢复正常。
综上所述,在心肌细胞中,缺氧后TRPV4被激活而引发钙离子内流,线粒体受损进而导致ROS 大量生成,进一步通过CAT 和SOD 调控细胞内氧化应激系统。本研究为心肌细胞抗缺氧药物的开发提供了一个新的靶点。
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
临床肺科杂志(2022年3期)2022-11-26
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国卒中杂志(2021年7期)2021-11-29
心肺血管病杂志(2020年5期)2021-01-14
教育教学论坛(2019年19期)2019-06-17
体育科学(2018年12期)2019-01-04
中国中药杂志(2017年11期)2017-06-22
科学中国人(2016年9期)2016-11-04
北方药学(2016年2期)2016-09-20
