
膜性肾病合并抗肾小球基底膜病2例并文献复习
2022-08-03 13:39邵晓琳石岩刘云马东红郭明好
河南医学研究 2022年14期
邵晓琳,石岩,刘云,马东红,郭明好
(新乡医学院第一附属医院 肾内科,河南 新乡 453100)
本文报道2例膜性肾病合并抗肾小球基底膜(glomerular basement membrane,GBM)病罕见病例。2例病例均为中年女性患者,均伴有急性起病、肉眼血尿、肾功能进行性下降、高血压、伴高滴度的抗GBM抗体等特征,及时行肾病理活检明确新月体性肾小球肾炎合并膜性肾病。积极给予血浆置换、激素冲击、丙种球蛋白、环磷酰胺治疗后随访观察至少1 a,2例患者的预后不同,本研究旨在报道引起不同预后的原因,为临床工作提供参考。
1 临床资料
患者1,女,51岁,因“间断双下肢水肿21个月,发现肾功能异常10 d”为主诉于2019年6月12日入院。21个月前无明显诱因出现双下肢水肿,就诊于新乡医学院第一附属医院,完善肾穿刺及相关辅助检查,确诊为“原发性肾病综合征Ⅰ期膜性肾病”,给予泼尼松30 mg(每日1次)联合他克莫司1 mg(每日2次)治疗,院外规律口服药物并逐渐减量至泼尼松 5 mg(隔日1次),他克莫司0.5 mg(每日早1次,晚上隔日1次),定期复查24 h尿蛋白,波动在0.7~1.9 g。10 d前无明显诱因出现发热,最高温度38.2 ℃,伴肉眼血尿,尿量约1 000 mL·d-1,无咳嗽、胸闷、气促,就诊于新乡医学院第一附属医院门诊,查尿常规:尿蛋白(+++),隐血(+++),红细胞851个·μL-1。生化:尿素16.12 mmol·L-1,肌酐158.7 μmol·L-1,尿酸465 μmol·L-1,白蛋白33.6 g·L-1。24 h尿蛋白3.1 g。建议住院治疗,患者拒绝。后就诊于鹤壁市第一人民医院,给予抗感染等治疗后未再发热,仍有肉眼血尿,且逐渐出现恶心、呕吐,复查血肌酐逐渐上升至600 μmol·L-1。为求进一步治疗再来新乡医学院第一附属医院,门诊以“肾病综合征Ⅰ期膜性肾病;急性肾损伤”收入肾内科。患者自发病来,神志清楚,精神差,饮食、睡眠差,大便正常,小便量约300 mL·d-1,体质量较前增加5 kg。既往史、个人史、家族史无特殊。入院查体:体温36.3 ℃,心率80次·min-1,呼吸20次·min-1,血压128/82 mmHg(1 mmHg=0.133 kPa),体质量66 kg,心肺腹查体无异常,双下肢重度凹陷性水肿。入院查血常规:白细胞11.7×109L-1,血红蛋白100 g·L-1。尿常规:尿蛋白(+++),隐血(+++),红细胞581个·μL-1。24 h尿蛋白5.9 g。血生化:肌酐669.6 μmol·L-1,尿酸245 μmol·L-1,尿素13.5 mmol·L-1,白蛋白25.4 g·L-1。抗GBM抗体148.23 RU·mL-1。血清抗磷脂酶A2受体抗体262.9 RU·mL-1。免疫五项:免疫球蛋白G 4.8 g·L-1,补体C4 0.43 g·L-1。超敏C反应蛋白94 mg·L-1。红细胞沉降率105 mm·h-1。肾与血管超声:左肾120 mm×41 mm,右肾125 mm×48 mm,双肾实质回声增强,集合系统光点弥散,与实质分界不清,肾静脉及动脉未见明显异常。胸部CT示双肺上叶炎症改变,右肺下叶结节。送检1条肾皮髓质组织,光镜下可见5个肾小球,其中3个肾小球细胞纤维性新月体形成,余肾小球体积大小正常。特殊染色可见上皮下嗜复红蛋白沉积,GBM增厚及较多量“钉突”形成。肾小管及间质病变中度,多灶性小管萎缩,间质炎细胞浸润及纤维化,小管腔内可见蛋白管型及红细胞管型。血管未见明显改变。肾小球毛细血管壁线性及颗粒样沉积,IgG(++),IgA(-),IgM(-),C3(+),C4(-),C1q(-),纤维蛋白相关抗原(-),乙型肝炎表面抗原(-),乙型肝炎核心抗原(-),IgG1(++),IgG2(-),IgG3(-),IgG4(++),Kappa(-),Lambda(++)。免疫组化结果示M型磷脂酶A2受体(phospholipase A2 receptor,PLA2R)阴性。电镜示肾小球脏层上皮细胞足突广泛融合,上皮下多数块状电子致密物沉积。肾小管上皮溶酶体增多,部分萎缩。肾间质淋巴单核细胞浸润伴胶原纤维增生。最终诊断:Ⅱ期膜性肾病合并Ⅰ型新月体性肾小球肾炎。
治疗上给予血浆置换2 400 mL·次-1,共11次。间断给予甲泼尼龙0.5 g·d-1,分别连用5、3、3 d静脉滴注,共11次,随后给予口服泼尼松片60 mg·d-1维持治疗,丙种球蛋白15 g·d-1,分别连用5、3、3 d,静脉滴注,共11次,间断环磷酰胺0.8 g·d-1,静脉滴注,共2次。患者入院第3天尿量减少至100 mL,给予14次血液透析肾替代治疗后尿量恢复,无尿期持续1个月。11次血浆置换后复查血肌酐134 μmol·L-1,抗GBM抗体34.62 RU·mL-1。院外口服泼尼松片50 mg·d-1,规律减量至目前10 mg·d-1维持治疗,院外起初每月给予环磷酰胺0.8 g静脉滴注,共6次,随后每3个月给予环磷酰胺0.8 g静脉滴注,共4次。随访1 a后复查抗GBM抗体、抗磷脂酶A2受体抗体正常,复查血肌酐118.5 μmol·L-1,尿蛋白/肌酐比值为0.51。随访2 a后复查血肌酐109.9 μmol·L-1,尿蛋白/肌酐比值为0.16。
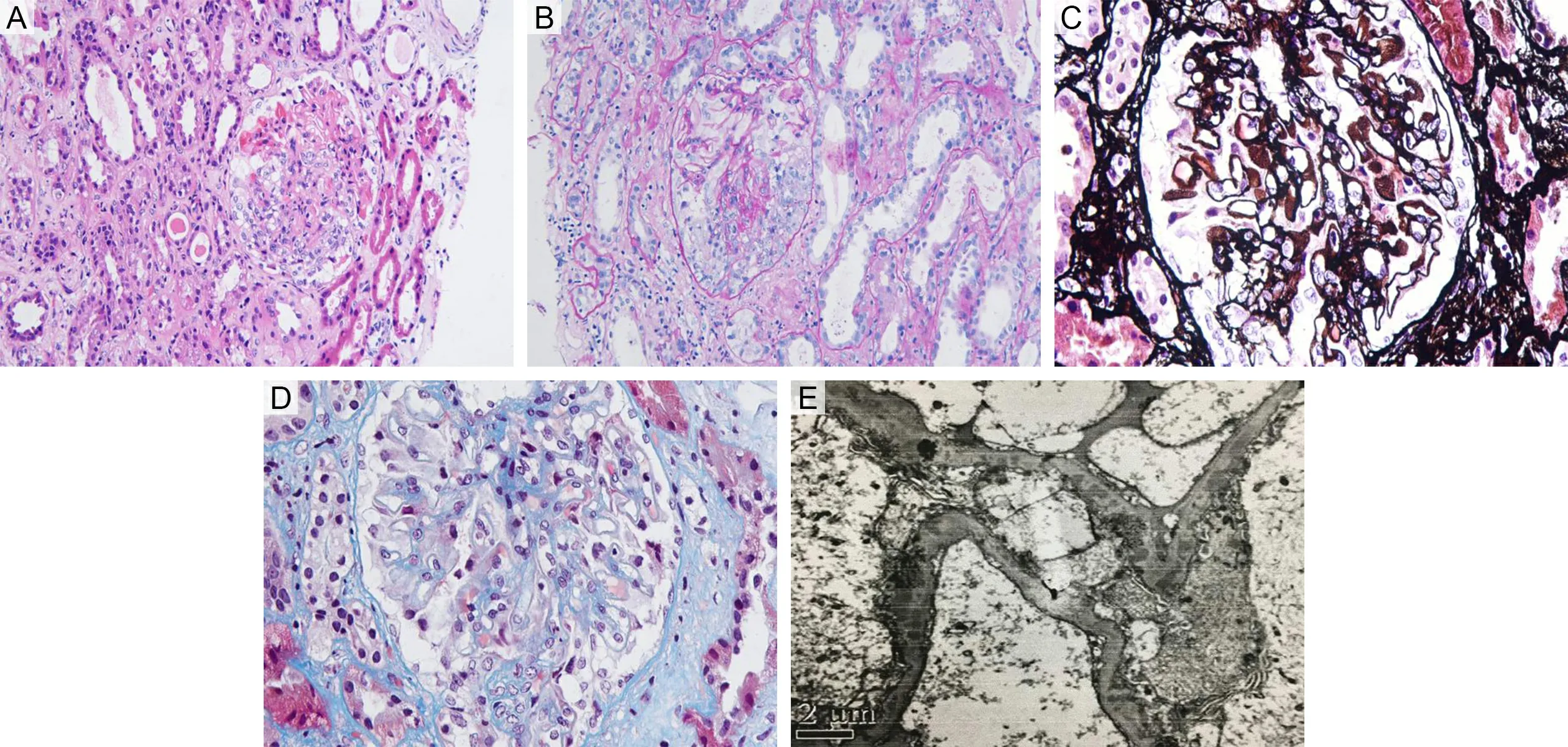
A为光镜下肾脏结构(HE染色×200);B为肾小球细胞性新月体(PAS染色×200);C为肾小球毛细血管袢节段坏死(PASM染色×400);D为肾小球细胞性新月体(Masson染色×400);E为肾小球脏层上皮细胞足突广泛融合,上皮下多数块状电子致密物沉积(电镜×8 000)。
患者2,女,46岁,因“双下肢水肿3个月余,发热9 d,胸闷3 d”于2020年8月11日收入院。3个月余前无明显诱因出现双下肢凹陷性水肿,进行性加重,伴双下肢胀痛,泡沫尿增多及腹胀,无肉眼血尿。至新乡医学院第一附属医院就诊,血常规示血红蛋白117 g·L-1。尿常规示蛋白(++),隐血(+)。尿蛋白/肌酐比值0.88。血生化示肌酐49 μmol·L-1,尿酸422 μmol·L-1,总胆固醇7.52 mmol·L-1,甘油三酯2.86 mmol·L-1,白蛋白29.2 g·L-1,超敏C反应蛋白0.36 mg·L-1。血管炎免疫4项、抗GBM抗体、血清抗磷脂酶A2受体抗体、抗核抗体、抗双链DNA抗体均阴性;凝血功能无异常。初步诊断为肾病综合征,建议行肾穿刺活检术明确病理类型,患者拒绝。院外给予昆仙胶囊治疗15 d,后自行停药。9 d前受凉后出现发热,最高体温38.7 ℃,伴寒战、畏寒、乏力、咳嗽、咳痰、头痛、咽痛,至当地医院接受抗感染治疗后未见好转。3 d前出现胸闷,夜间不能平卧,尿量减少呈洗肉水样,伴发热、腹胀、咳嗽、咳痰,至新乡医学院第一附属医院门诊就诊,生化示肌酐254.4 μmol·L-1,白蛋白23.3 g·L-1;胸部CT提示两肺慢性炎症改变,双侧胸腔积液,甲状腺病变。门诊以“肾病综合征;发热待查”收入肾内科,自发病来,小便减少至300 mL,体质量较前增加2 kg。入院查血常规:白细胞计数10.2×109L-1,血红蛋白78 g·L-1。尿常规:蛋白(++),隐血(+++),红细胞400个·μL-1。24 h尿蛋白定量2.8 g。血生化:肌酐645.6 μmol·L-1,尿酸520 μmol·L-1,总胆固醇4.22 mmol·L-1,甘油三酯1.24 mmol·L-1,白蛋白27 g·L-1。血管炎免疫4项正常。抗GBM抗体284.66 RU·mL-1,血清抗磷脂酶A2受体抗体75.0 RU·mL-1,免疫球蛋白G 4.7 g·L-1,超敏C反应蛋白238.40 mg·L-1,红细胞沉降率85 mm·h-1,纤维蛋白原7.88 g·L-1,纤维蛋白降解产物39.1 mg·L-1,D-二聚体16.7 mg·L-1。泌尿系彩超示左肾切面内径125 mm×55 mm,右肾切面内径134 mm×53 mm,实质回声增强。胸部CT示两肺炎症病变,两侧胸腔积液,提示贫血、心包积液及甲状腺病变。再次与患者及家属沟通行肾穿刺检查,患者及家属同意,遂行肾穿刺活检术。送检2条肾皮髓质组织,光镜下可见4个肾小球,其中2个肾小球细胞纤维性新月体形成,余肾小球未见明显改变。特殊染色可见上皮下少量嗜复红蛋白沉积及GBM欠光滑。肾小管及间质病变轻-中度,多灶性小管上皮细胞空泡变性、脱落及间质炎细胞浸润,小管腔内可见蛋白管型。血管未见明显改变。肾小球毛细血管壁线性及颗粒样沉积,IgG(++),IgA(-),IgM(-),C3(-),C4(-),C1q(-),FRA(-),乙型肝炎表面抗原(-),乙型肝炎核心抗原(-),IgG1(++),IgG2(-),IgG3(-),IgG4(-),Kappa(-),Lambda(弱阳性)。免疫组化示PLA2R阴性。电镜示肾小球系膜细胞和基质轻度增生,基底膜节段增厚伴钉突样增生,上皮下、基底膜内电子致密物沉积,上皮细胞足突广泛融合。肾小管上皮细胞空泡变性伴溶酶体增多。肾间质无明显病变。最终诊断为Ⅰ型新月体性肾小球肾炎合并膜性肾病。
治疗上依次给予甲泼尼龙0.5 g·d-1,静脉滴注3 d,地塞米松磷酸钠注射液7.5 mg·d-1,静脉注射7 d,甲泼尼龙0.5 g·d-1,静脉滴注3 d。6次甲泼尼龙治疗后调整为6 mg·d-1静脉注射,2周后再次调整为5 mg·d-1静脉注射,出院时调整为口服泼尼松30 mg·d-1,因依从性较差在1 a内自行减量至5 mg·d-1。因肾功能较差间断静脉滴注环磷酰胺0.4 g·d-1,共2次,静脉滴注丙种球蛋白15 g·d-1,共3次,血浆置换2 400 mL·次-1,共10次,每周血液透析2~3次,10次血浆置换后复查血肌酐458.1 μmol·L-1,抗GBM抗体76.36 RU·mL-1。患者院外口服环磷酰胺片50 mg,每日2次,共0.7 g。随访1个月后复查血肌酐274.5 μmol·L-1,抗GBM抗体66.50 RU·mL-1,尿蛋白/肌酐比值为5.17,尿量恢复至1 000 mL时停血液透析,无尿期持续3个月。此后间断3次静脉滴注环磷酰胺(每次0.8 g),随访1 a后复查尿蛋白/肌酐比值为2.57,肌酐213.7 μmol·L-1,白蛋白35.8 g·L-1,抗GBM抗体67.65 RU·mL-1。
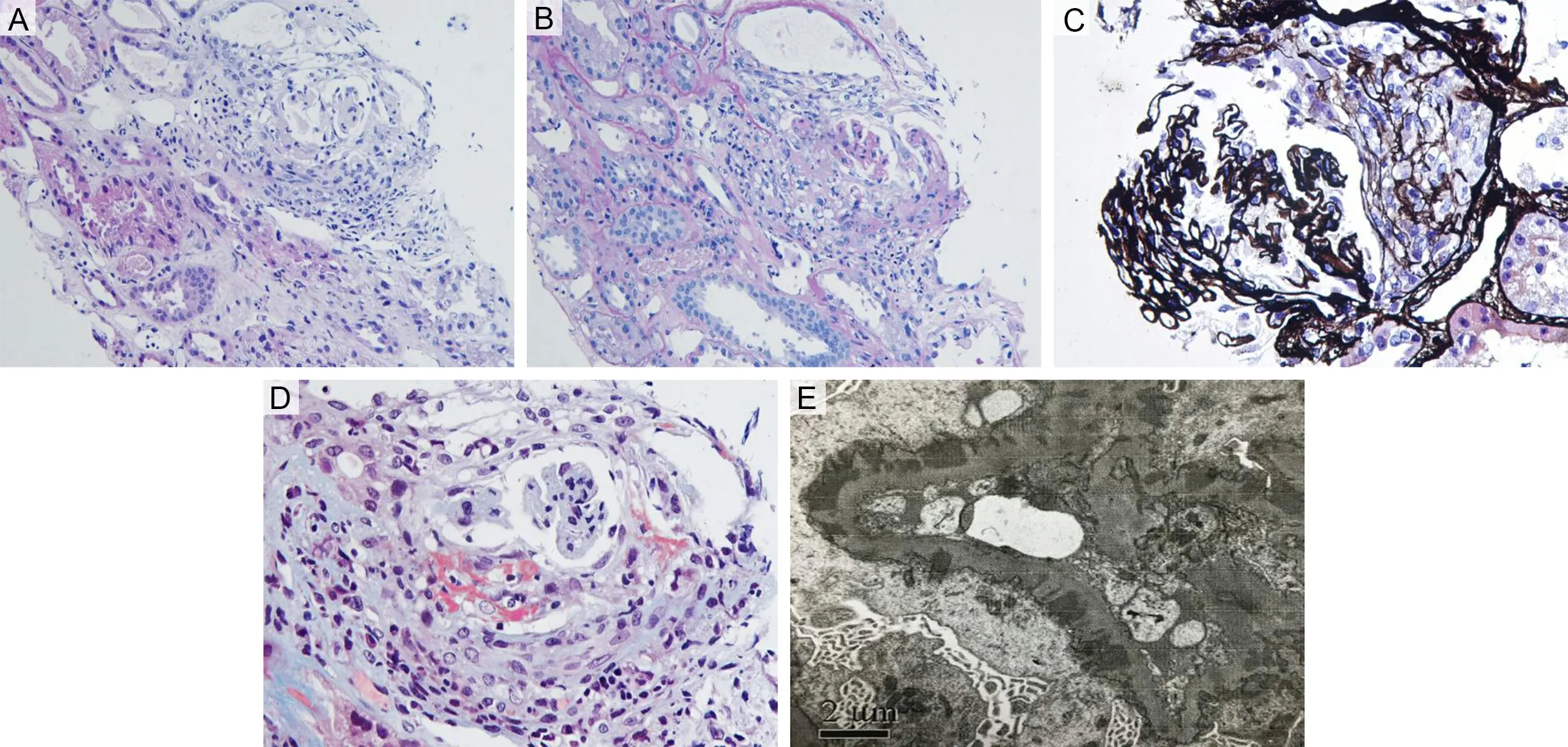
A为光镜下肾脏结构(HE染色×200);B为肾小球细胞性新月体(PAS染色×200);C为肾小球毛细血管袢节段坏死(PASM染色×400);D为肾小球细胞性新月体(Masson染色×400);E为上皮下、基底膜内电子致密物沉积,上皮细胞足突广泛融合(电镜×10 000)。
2 讨论
膜性肾病是一种自身免疫性疾病,是非糖尿病成人肾病综合征的主要原因,很少出现快速进展性肾衰竭和血尿。综合分析2例患者的病情,可分为两阶段,第一阶段临床表现为肾病综合征,第二阶段出现肉眼血尿,肾功能快速进展,伴高滴度的抗GBM抗体,临床表现为急进性肾炎综合征、肾病综合征;2例患者都由肾病综合征发展成合并急进性肾炎综合征,通过及时肾活检明确了病理诊断,为治疗提供了依据,积极给予血浆置换、激素冲击、丙种球蛋白、环磷酰胺治疗,期间给予短暂血液透析肾替代治疗直至尿量恢复。
原发性膜性肾病合并抗GBM抗体病并不常见,确切的发病机制尚不清楚,有推测可能是由于膜沉积改变GBM并导致内源性GBM抗原释放到循环中,从而导致抗GBM抗体的形成[1]。作为膜性肾病的病因,M型磷脂酶A2受体抗原的释放可能导致了GBM的构象改变,从而引起抗GBM肾炎[2]。目前国内外关于膜性肾病合并抗GBM病的病例以中老年为主,多表现为肾功能不全,肉眼血尿、大量蛋白尿发生率高[3]。有文献报道抗GBM病首发症状主要为发热(42.5%)、食欲减退(23.4%)、肉眼血尿(17.0%),主要并发症为肺部感染(17.0%)、心力衰竭(17.0%)[4]。本文2例患者首发症状均是发热,后出现肉眼血尿、食欲减退,期间合并肺部感染、心力衰竭症状,与文献报道一致[4]。
2例患者肾活检中均有纤维性新月体形成。据文献报道,新月体在膜性肾病中极为罕见,患病率为0.26%~0.39%[5],如果肾病理提示新月体肾炎,考虑叠加其他疾病。2例患者活检均确定叠加Ⅰ型新月体性肾小球肾炎。2例患者肾活检肾小球数量不多,病理的可靠程度有待商酌,从病理上看病例2并不比病例1重,但无尿期持续时间几乎是病例1的2倍,因此病例2的病理并未反映真实情况。原发性膜性肾病中被自身抗体识别的2种主要抗原是PLA2R和血小板反应蛋白1型结构域7A,分别占比70%~80%和3%~5%,普遍存在的IgG亚类是IgG4[6]。在抗GBM疾病中,绝大多数患者是由自身抗体识别Ⅳ型胶原a3链非胶原区的靶抗原,其主要的IgG是IgG1和IgG3,可以激活补体途径,更罕见的是IgG4或IgA[7]。有研究显示,特发性膜性肾病患者肾组织IgG4阳性为81.48%[8],本文病例1患者IgG1(++),IgG4(++),病例2患者IgG1(++),IgG4(-)。2例患者血清抗PLA2R抗体均阳性,肾脏病理免疫组化显示PLA2R均为阴性。有文献报道,肾PLA2R抗原在特发性膜性肾病患者中可能是假阴性的,因为抗原抗体沉积可以随着抗原性的消失而清除[9]。
有文献报道,抗GBM肾炎与膜性肾病的先后顺序不同,预后不同;中老年患者多以肾小球膜性肾病起病,肾脏表现以水肿为主,预后差;青年患者多以抗GBM肾炎起病,肾脏表现均为血尿,预后好;16~71岁患者抗GBM肾炎与肾小球膜性肾病同时发病,表现为血尿或水肿为主,约50%患者进入终末期肾病或死亡,约50%患者治愈[10]。本文2例患者为中老年患者,均出现膜性肾病合并抗GBM肾炎,以水肿为主,据文献报道考虑患者预后较差,但经过积极治疗后,并未进入到长期肾脏替代治疗或死亡,经过规范治疗的病例1预后更好。有研究显示,抗GBM病合并膜性肾病患者肾脏预后较好,约62.5%的患者肾功能恢复较好,不依赖于透析治疗[11],与本文病例一致。
2例患者经肾活检明确诊断后给予激素、环磷酰胺治疗,随访1 a后病例1复查血肌酐、尿蛋白较前好转,抗GBM抗体、血清抗磷脂酶A2受体抗体均转为阴性。病例2血肌酐、尿蛋白较前稍好转,抗GBM抗体仍为阳性。预后不同的原因可能为病例2的初始抗GBM抗体滴度较高,无尿期时间较长,依从性较差,且病例2住院治疗期间出现较重的肺部感染、胃肠道不适等不良反应,环磷酰胺的剂量较病例1减半,院外也未规律使用。贾晓玉等[12]通过临床研究发现,循环中的抗GBM抗体是否可以转阴主要取决于抗体的初始滴度,转阴患者初始抗体滴度低于未转阴患者。2021改善全球肾脏病预后组织指南推荐使用糖皮质激素和环磷酰胺作为初始免疫抑制治疗抗GBM肾炎[13],可见免疫抑制剂在治疗过程中的重要地位。
膜性肾病出现快速进展性肾功能衰竭应高度警惕合并其他疾病的可能,应及时进行肾活检明确诊断,并积极治疗。2例患者均急性起病,伴肉眼血尿、肾功能进行性下降、高血压、高滴度的抗GBM抗体,及时接受肾脏活检,明确新月体性肾小球肾炎合并膜性肾病。2例患者肾活检提示血清抗PLA2R抗体和肾PLA2R抗原并不一致,可能是因为抗原抗体沉积随着抗原性的消失而清除。2例患者预后不同的原因可能与初始抗GBM抗体滴度较高、无尿期时间较长、依从性较差有关,为以后此类患者的治疗提供了经验。在治疗上应积极给予甲泼尼龙、环磷酰胺冲击治疗以及血浆置换以改善肾功能,摆脱肾脏替代治疗。
猜你喜欢
基层中医药(2022年3期)2022-07-22
现代临床医学(2022年2期)2022-04-19
健康体检与管理(2022年2期)2022-04-15
临床荟萃(2021年4期)2021-12-24
祝您健康·文摘版(2021年12期)2021-12-08
婚育与健康(2021年3期)2021-07-05
健康之家(2021年19期)2021-05-23
中华临床免疫和变态反应杂志(2020年4期)2020-12-08
健康必读(上旬刊)(2019年12期)2019-10-21
祝您健康(2000年11期)2000-12-31
