
绒毛崖豆藤中一种查尔酮提取物的体内外代谢及药代动力学研究
2022-08-01 08:27倪恒凡
天然产物研究与开发 2022年7期
倪恒凡,刘 玲,万 丽*
1四川大学华西医院药剂科,成都 611137;2成都中医药大学药学院,成都 610041
天然化合物对新药的发现做出了巨大贡献,黄酮类化合物是天然产物中的一大类[1],因为其具有良好的抗炎、抗肿瘤、抗菌、抗溃疡、保肝、保护心血管等活性备受大家关注[2-9],从豆科崖豆藤属植物绒毛崖豆藤(MillettiavelutinaDunn)茎中提取分离出的抗炎活性化合物2,5-二甲氧基呋喃[4″,5″:3,4]查耳酮(2,5-dimethoxyfuran[4″,5″:3,4]chalcone,1)属于查尔酮类化合物。Ma等[10]研究结果显示,该化合物具有较强的抗炎活性,其在体外细胞实验中主要通过抑制IL-1β分泌和Caspase-1激活,并抑制ASC齐聚,从而表现出良好的抗炎活性。并在小鼠体内实验中可明显缓解LPS诱导的小鼠急性休克。在新药开发的早期阶段,进行药物代谢的相关研究有助于快速识别安全、有效、药代动力学性质良好的化合物,提前淘汰不合格的化合物,节约研发成本,目前尚未在国内外期刊上发现对该化合物的体内外代谢数据研究报道。本研究通过对2,5-二甲氧基呋喃[4″,5″:3,4]查耳酮在大鼠、小鼠、恒河猴、Beagle犬和人5个种属肝微粒体和大鼠体内的代谢产物研究,并通过对2,5-二甲氧基呋喃[4″,5″:3,4]查耳酮及其代谢产物(M1)体内外药代动力学研究探究该化合物的主要物质作用基础,为其进一步的机制研究奠定基础。
1 材料与方法
1.1 材料
美国Thermo公司Q Exactive高分辨质谱(含ESI源,软件Xcalibur3.2、Compound Discover 2.0);美国AB SCIEX公司QTRAP5500三重四级杆质谱仪(软件Analyst 1.6.2、MμLtiQuant 3.3);日本Shimadzu公司超高速液相色谱,美国Waters公司ACQUITY UPLC®BEH C18色谱柱。
样品1(自制,No:20190221,纯度≥98%,结构见图1),ESI-MS:m/z309.122 1(calcd for C19H16O4,309.112 7)[M+H]+[10]。样品M1(自制,No:20190311,纯度≥98%,结构见图1),ESI-MS:m/z311.1277(alcd for C19H18O4,311.1283)[M+H]+。内标辛二酰苯胺异羟肟酸(大连美仑生物技术有限公司,No:00502A,纯度≥98%)。人种属的肝微粒体(20 mg/mL)、大鼠种属的肝微粒(20 mg/mL)、比格犬种属的肝微粒体(20 mg/mL)、猴种属的肝微粒体(20 mg/mL)、小鼠种属的肝微粒体(20 mg/mL),NADPH反应启动液(武汉普莱特生物医药技术有限公司),于-80 ℃冷冻保存。甲醇、乙腈(色谱纯,Sigma-Aldrich公司)。甲酸(色谱纯,FlukaAnalytical公司)。
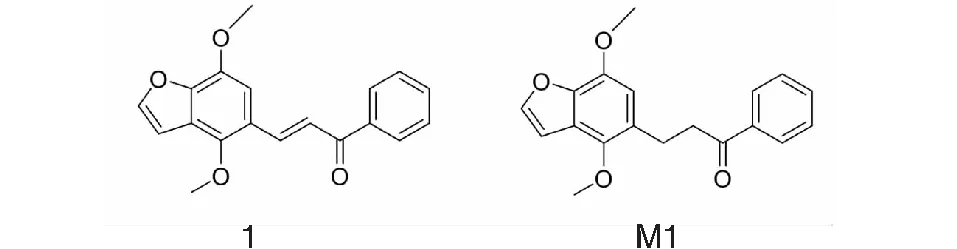
图1 样品1及M1结构式Fig.1 Structures of sample 1 and M1
实验动物:SPF级SD雄性大鼠,体重200±10 g,SPF级C57雄性小鼠,体重22±2 g均购买于成都达硕实验动物有限公司,许可证号:SCXK(川)2014-028。
1.2 溶液的配制
1.2.1 给药溶液的配制
样品1注射液制备方法:精密称取样品1适量,加入生理盐水适量,制备成浓度1 mg/mL,于4 ℃冰箱密封保存,备用。
1.2.2 储备液的配制
精密称取样品1及M1适量,用甲醇溶解制备成质量浓度为1 mg/mL的储备液,于4 ℃冰箱密封保存,备用。
1.2.3 内标溶液的配制
精密称取SAHA对照品适量,用乙腈稀释成20 ng/mL的内标工作液。
1.3 LC-MS/MS条件
1.3.1 液相条件
QTRAP5500三重四级杆质谱:ACQUITY UPLC®BEH C18色谱柱(2.1 mm×50 mm,1.7 μm),流动相为:0.1%甲酸水(A相),乙腈(B相)梯度洗脱(0~1 min,10%→90% B,1~3 min,90% B),流速为0.5 mL/min,柱温35 ℃,平衡1.5 min,进样体积1 μL。
Q Exactive高分辨质谱:ACQUITY UPLC®BEH C18色谱柱(2.1 mm×50 mm,1.7 μm),流动相为:0.1%甲酸水(A相),乙腈(B相)梯度洗脱(0~15 min,10%→90% B,15~20 min,90% B),流速为0.5 mL/min,柱温35 ℃,平衡1.5 min,进样体积2 μL。
1.3.2 质谱条件
QTRAP5500三重四级杆质谱:采用MRM(多反应检测模式)检测,质谱条件为:ESI+(正离子模式),样品1监测离子对为:m/z309.1→m/z267.1,碰撞能量为17.73 V;M1监测离子对为:m/z311.1→m/z191.1,碰撞能量为15.27 V;内标SAHA监测离子对为:m/z265.1→m/z232.1,碰撞能量为20 V;去簇电压(DP)均为100 V;离子束聚焦电压(EP):10 V;碰撞池出口电压(CXP):10 V;驻留时间为0.10 s。
Q Exactive高分辨质谱:质谱条件为:ESI+(正离子模式),喷雾电压(IS):4 500 V;雾化气压力(GS1):50 Psi;气帘气压力(CUR):15 Psi;辅助气压力(GS2):45 Psi;离子源温度(TEMP):550 ℃;簇裂解电压(DP):55 V;碰撞能量(CE):35 V。
1.4 样品1在各种属代谢产物研究
配制孵育体系为200 μL(PBS 188 μL、NADPH 12 μL),再加入样品12 μL(2 mmol/L),恒温水浴预孵育(37 ℃、5 min),再加入各种属的肝微粒体5 μL(20 mg/mL)启动反应。在37 ℃水浴锅中继续孵育60 min后加入400 μL含SAHA(20 ng/mL)的冰乙腈终止反应。涡旋3 min,离心15 min(13 000 r/min),取上清液进样,实验平行3份[11]。样品进UPLC-QE-Orbitrap-MS检测,通过对比孵育0 min和60 min样品离子流图,寻找样品1在各种属肝微粒体中的代谢产物。
1.5 样品1在C57小鼠体内代谢产物研究
随机取C57小鼠分为对照组、血浆组和粪尿组,每组3只。将小鼠放置在代谢笼中,给药前12 h禁食不禁水,对照组尾静脉注射生理盐水,实验组注射样品1(10 mg/kg)。血浆组分别于给药后0.5、1.5 h收集血液样品,尿液和粪便样品于给药后收集48 h内的粪便和尿液(每8 h收集一次)。取出离心转速为3 500 r/min,时间15 min,后取上清液即得血浆样品,样品保存于低温冰箱-80 °C。样品预处理如下:
血浆样品:取0.1 mL血浆样品加入到预先活化的SPE小柱中,用1 mL 10%甲醇洗涤3次,弃去,再加入1 mL甲醇,将洗脱液进行氮吹,后加100 μL甲醇复溶,13 000 r/min离心15 min,取上层清液进样分析。
尿液样品:取0.5 mL尿液样品加入到预先活化的SPE小柱中,用1 mL 10%甲醇洗涤3次,弃去,再加入1 mL甲醇,将洗脱液进行氮吹,后加100 μL甲醇复溶,13 000 r/min离心15 min,取上层清液进样分析。
粪便样品:将粪便干燥后研磨粉碎,每0.1 g粪便粉末加入2 mL醇浸提12 h后再超声15 min,再取出3 500 r/min离心15 min,取上层清液于37 ℃进行氮吹,所得残渣加入100 μL醇复溶,3 500 r/min离心15 min,取上层清液进样分析。
1.6 样品1及M1在大鼠体内的药代动力学研究
经过“1.3”项研究,在5个种属的肝微粒体的孵育结果中都找到了样品1的代谢产物样品1加氢还原物(M1),经过下文中的二级质谱确证后,推断出样品1还原物的结构,然后本课题组合成了该代谢产物,并在大鼠样品1静脉注射后进行样品1及M1的定量研究并计算药代动力学相关参数。
1.6.1 大鼠颈静脉插管手术
大鼠用10%水合氯醛(3.5 mL/kg)进行麻醉处理,麻醉后固定于无菌操作台中,剔除左边颈部的毛发后,用酒精擦拭干净皮毛,待大鼠麻醉后,用手术剪剪开皮肤找到颈静脉,使用1 mm外径0.5 mm内径的聚乙烯管插入颈静脉中缝合固定,后将聚乙烯管从后颈部穿出固定于耳后,缝合后将大鼠静置24 h,禁食不禁水。
1.6.2 血浆样品收集
将6只SD大鼠从颈静脉注射给药样品1,给药剂量为10 mg/kg,分别于给药前和给药后0.083、0.25、0.5、1、2、4、8、10、24 h,从颈静脉插管中取血0.2 mL到EP管中,3 500 r/min离心15 min,分离出上层血浆冷藏于-40 ℃待用。
1.6.3 血浆样品处理
取大鼠血浆30 μL于1.5 mL的EP管中,加入含20 ng/mL内标SAHA的乙腈120 μL涡旋30 s后13 000 r/min离心15 min,取上层离心液80 μL装入进样瓶中进UFLC-MS/MS分析。
1.7 方法学考察
样品1及M1方法学验证按照《药物非临床药代动力学研究技术指导原则》进行方法学验证[12],主要验证内容有专属性、线性范围、精密度与准确度、基质效应、稳定性等。
2 实验结果
2.1 方法学验证
2.1.1 专属性
按照“1.6.3”项下条件,取空白大鼠的血浆30 μL,分别制备空白样品和含内标和样品1标准溶液的对照品及含内标和M1标准液的对照品,与实验血浆样品进行UFLC-MS/MS分析,进行样品的专属性考察。结果显示:空白样品无干扰峰,内标SAHA与样品1及M1分离度好,保留时间分别为1.5、1.99及1.96 min,证明该方法专属性良好,特异性高(见图2)。

图2 样品1、M1分别与内标SAHA的色谱图Fig.2 Chromatograms of compound 1,M1 and internal standard SAHA注:空白血浆样品(1);血浆中分别加入样品1对照品或M1对照品和SAHA(2);给药后血浆样品(3)。a:M1;b:样品1;c:SAHA内标。Note:Blank plasma sample (1);Sample 1 or M1 and SAHA were added to the plasma (2);Plasma sample after administration (3).a:M1;b:Sample 1;c:SAHA internal standard.
2.1.2 线性与范围
于空白的大鼠血浆中分别加入系列浓度的样品1、M1标准溶液,终浓度配制成分别为3、10、30、100、300、1 000 ng/mL的标准品溶液,进行UFLC-MS/MS分析,将药物与内标峰面积比值与药物终浓度作线性回归,以加权最小二乘法(权重系数为1/C2),分别考察三条标准曲线,样品1的结果分别为:y=0.012 04x-0.001 04(R=0.997 01),y=0.0127x+ 0.002 40(R=0.991 30),y=0.012 55x-0.009 86(R=0.994 63),表明样品1质量浓度在3~1 000 ng/mL范围内成线性相关,根据信噪比S/N≥10得到最低定量限为3 ng/mL。M1考察三条标准曲线结果分别为:y=0.012 36x-0.006 06(R=0.997 77),y=0.012 21x+0.002 29(R=0.993 60),y=0.011 63x-0.000 04(R=0.996 08),根据信噪比S/N≥10得到最低定量限为3 ng/mL。
2.1.3 精密度与准确度
按照“1.6.3”项下方法,分别配制样品1及M1质量浓度为9、100、800 ng/mL的低、中、高质量浓度的质控样品,每个质量浓度质控样品平行6份,进样分析,测定样品1及M1的浓度,考察日内精密度;连续进样3天,考察日间精密度;同法制备该质量浓度质控样品,进样分析,计算各质控样品测得浓度与理论浓度的回收率,评价该方法的准确度;由表1可知,低、中、高质量浓度质控样品的日内、日间RSD均小于10%,样品1的回收率在100.96%~103.21%,M1的回收率在99.20%~105.83%,符合生物样品定量分析的相关要求[13]。表明该方法精密度、准确度良好,测得结果准确可信。
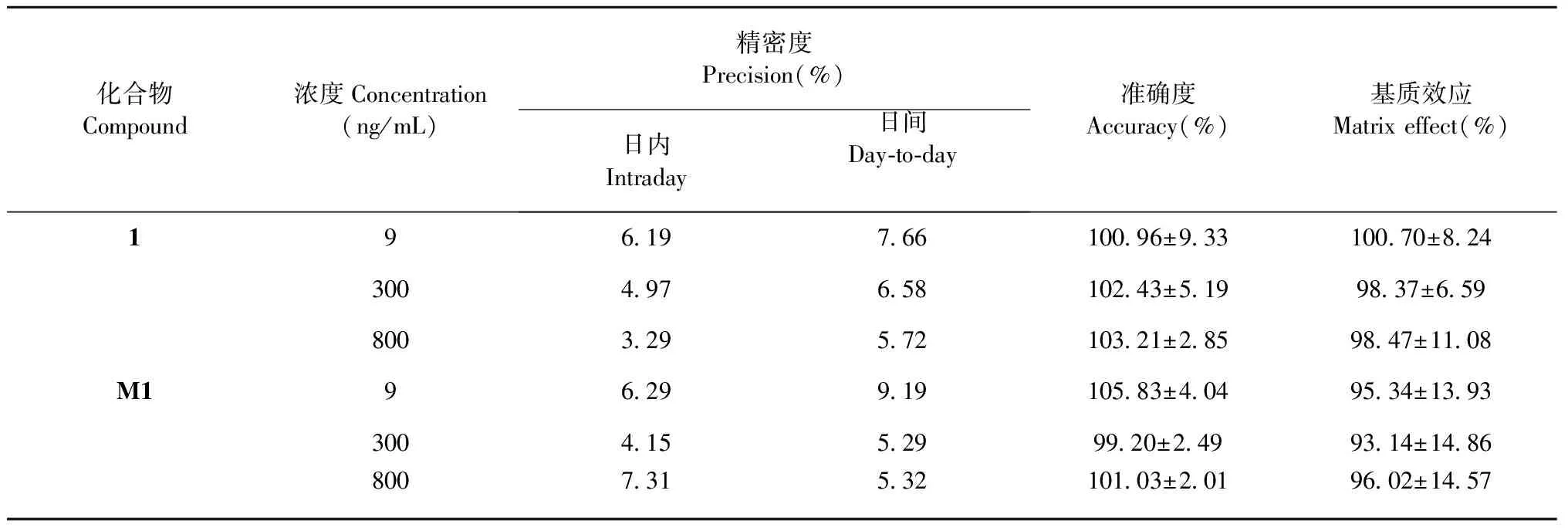
表1 样品1及M1的精密度、准确度和基质效应测定(n=6)
2.1.4 基质效应
按照“1.6.3”项下方法,用大鼠空白血浆配制质量浓度分别为9、300、800 ng/mL的低、中、高质量浓度的质控样品,同时用流动相分别配制相同质量浓度的样品1对照品溶液,进样分析,分别得峰面积A和B。每组平行5份。基质效应=A/B×100%。由表1结果显示,样品1基质效应在98.37%~100.70%,M1基质效应在93.14%~96.02%,偏差小于15%,表明此提取方法无基质效应。
2.1.5 稳定性
按照“1.6.3”项下方法,配制质量浓度分别为9、300、800 ng/mL的低、中、高质量浓度的质控样品,进样分析。每组平行5份,考察各质控样品于自动进样室内放置24 h的稳定性,-80 ℃放置15天的长期稳定性,以及冻融5次后的冻融稳定性。表2结果显示在上述各条件下,样品1及M1样品稳定性良好。
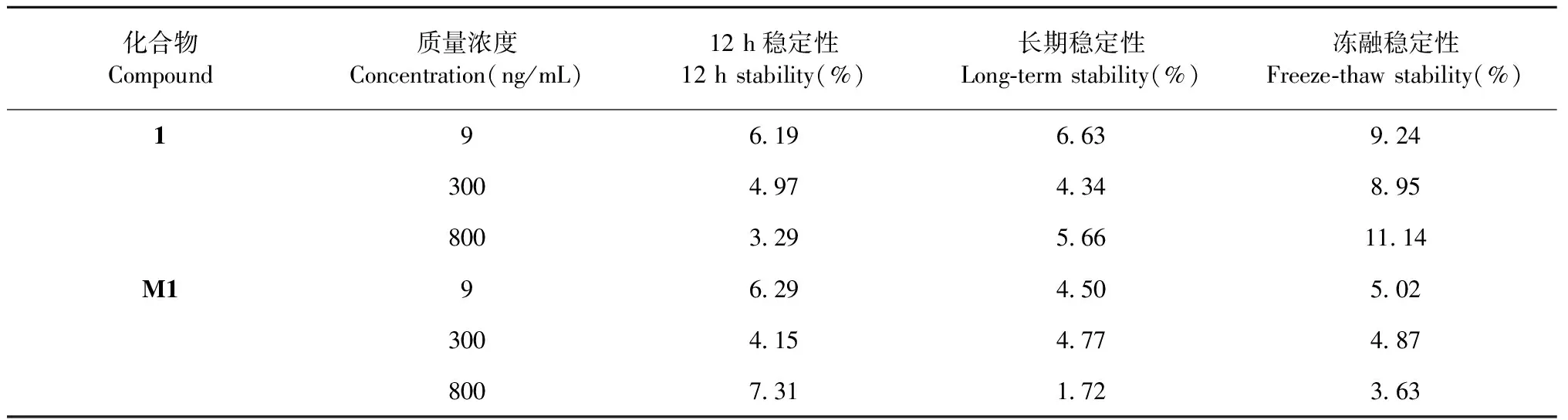
表2 样品1及M1的稳定性测定(n=5)
2.2 样品1在不同种属肝微粒体中的代谢产物
采用一级全扫描正离子方式检测(MS scan),比较0、60 min两个时间点的色谱图差异,初步判断样品1的体外代谢产物。结果显示5个种属均有2个体外代谢产物:M1(8.36 min),m/z311.127 7([M+H]+);M2(4.06 min),m/z299.128 3([M+H]+)(见图3)。由于体外代谢产物M1、M2与下文体内代谢产物一致,故在下文体内代谢产物中进行详细结构分析。
2.3 样品1在小鼠体内的代谢产物
将处理后的小鼠的血浆、尿液、粪便样本进UPLC-QE-Orbitrap-MS进行分析,通过比较给药前后的质谱图谱,结合各组分的保留时间、精确的一级母离子分子质量、碎片的分子质量等信息,推断出各个代谢产物的可能结构,预测代谢途径,在血浆样本中鉴定出原型药样品1及代谢产物M1、M2、M5共4个,在粪便样本中鉴定出原型药及代谢产物M1、M2、M3、M4、M5共6个,尿液样本中鉴定出原型药及代谢产物M1、M3、M4共4个,各个代谢产物保留时间详见下图4中粪便样本的总离子流图,各个代谢产物及原药结构见图5,各个代谢产物的二级碎片信息见表3。

图3 以人肝微粒体为代表的体外代谢产物总离子流图Fig.3 Total ion chromatogram of in vitro metabolites represented by human liver microsomes
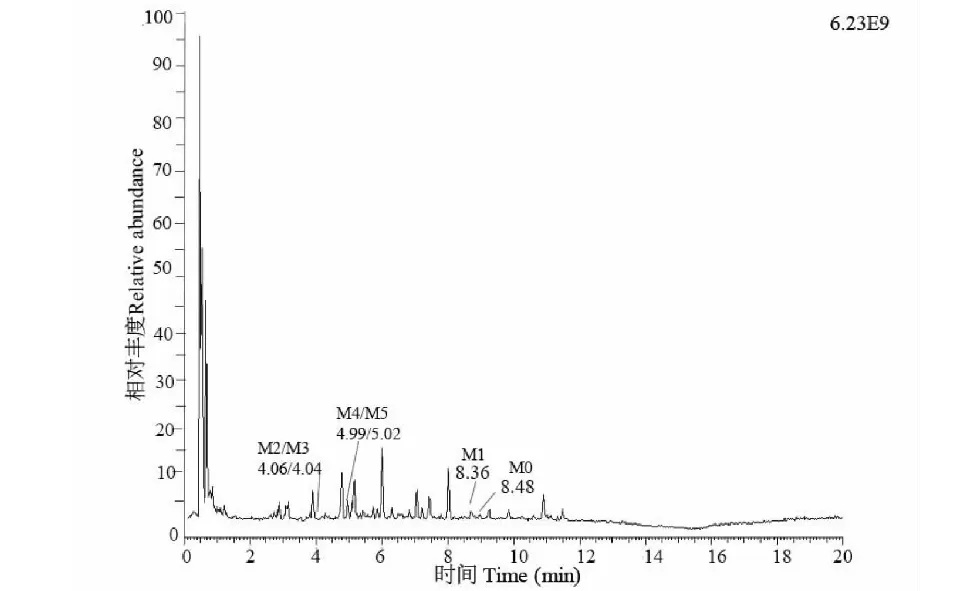
图4 以粪便为代表的体内代谢产物总离子流图Fig.4 Total ion current diagram of metabolites in the body represented by feces
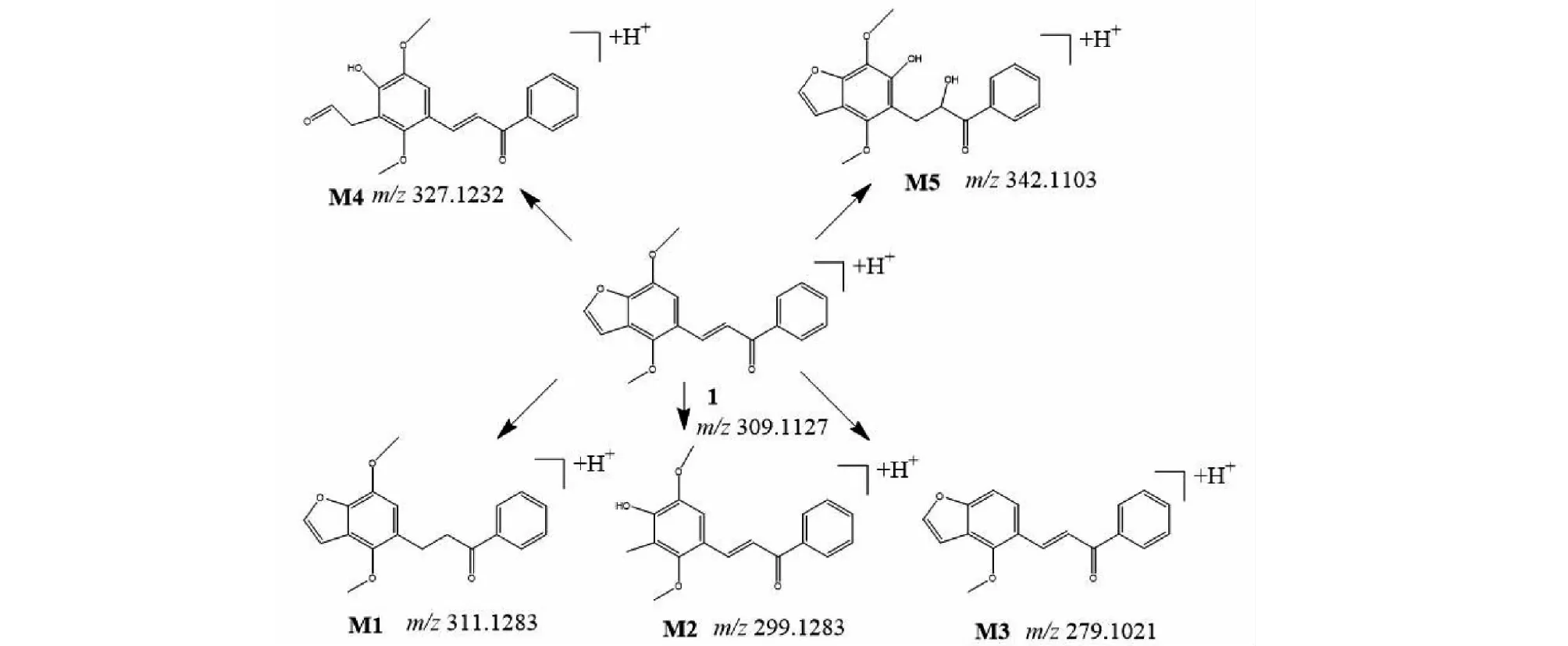
图5 样品1及代谢产物产物结构图Fig.5 Structure of compound 1 and metabolite products

表3 样品1代谢产物表征
如图4所示,样品1(C19H16O4)洗脱时间为8.48 min,准分子离子峰为[M+H]+m/z309.112 7(见图6a),其二级质谱主要的特征碎片离子有m/z267.101 6 [M-C2HO]+、179.074 0 [M+H-C9H6O]+、131.049 1 [M-C10H9O3]+、105.115 5 [M-C12H11O3]+(见图7a);可能的裂解途径见图8。利用代谢产物与原型药具有相似的断裂途径,m/z131.049 1、178.063 0、105.115 5可作为快速鉴别样品1体内代谢产物的特征性离子。
M1的准分子离子峰为[M+H]+m/z311.1277(图6b),推测其分子式为C19H18O4,比化合物1分子质量多2 Da,推断为化合物1加上两个H而成。其二级质谱主要的特征碎片离子(见图9)有m/z269.107 6、191.070 3、105.033 5。其中m/z269.107 6为M1去掉左侧呋喃环结构,比化合物1的原药碎片m/z267.1016多2 Da,其中m/z191.070 3为M1左侧的苯环和呋喃环的结构,m/z105.033 5为M1右侧的苯环外连接一个羰基的结构,从两个碎片上看还原所加的两个H原子未加到两侧的苯环、呋喃环及羰基上,由上推测M1可能为化合物1的中间双键加氢还原代谢产物,后经合成M1后确证。
M2的准分子离子峰为[M+H]+m/z299.127 3(见图6c),推测其分子式为C18H18O4,比化合物1分子质量少了10 Da,推断为化合物1结构中呋喃环打开后掉甲基而形成。其二级质谱主要的特征碎片离子(见图10)有m/z267.101 6、105.033 5。其中m/z267.101 6为M2去掉左侧的呋喃环的结构,m/z105.033 5为M2右侧的苯环外连接一个羰基的结构,掉的甲基确认在呋喃环上,由上推测M2可能为化合物1的呋喃环打开掉甲基形成。
M3的准分子离子峰为[M+H]+m/z279.101 1(见图6d),推测其分子式为C18H14O3,比化合物1分子质量少了30 Da,推断为化合物1结构中去掉甲氧基而形成。其二级质谱主要的特征碎片离子(见图11)有m/z251.106 7、201.055 1、105.034 0。其中m/z251.106 7碎片可能为M3去掉左侧的呋喃环打开后去掉甲基及O原子的结构,m/z201.055 1碎片可能为M3去掉右侧苯环的结构,m/z105.033 5为M3右侧的苯环外连接一个羰基的结构,由上推测M3可能为化合物1去甲氧基形成。
M4的准分子离子峰为[M+H]+m/z327.122 2(见图6e),推测其分子式为C19H18O5,比化合物1分子质量多了18 Da,推断为化合物1结构中加O及2个H原子而形成。其二级质谱主要的特征碎片离子(见图12)有m/z309.112 2、267.101 3、131.049 4、105.034 0。其中m/z309.112 2碎片可能为M4去掉左侧的呋喃环打开后去掉O原子形成,m/z267.101 3碎片可能为M4去掉左侧苯环的结构醛基,m/z105.033 5为M4右侧的苯环外连接一个羰基的结构,由上推测M4可能为化合物1呋喃环开环形成。
图6 化合物1及代谢产物的一级色谱图Fig.6 The MS spectra of 1 and metabolites

图7 化合物1及代谢产物的二级色谱图Fig.7 The MS/MS spectra of 1 and metabolites
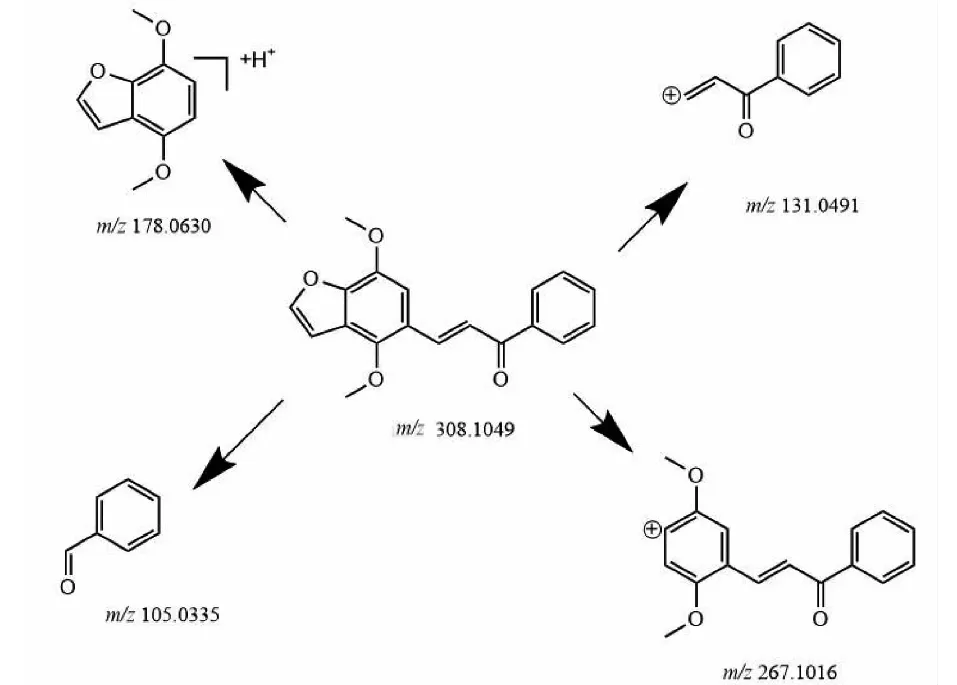
图8 样品1可能的裂解机制Fig.8 Possible fragmentation pathways of compound 1
M5的准分子离子峰为[M+H]+m/z343.118 1(图6f),推测其分子式为C19H18O6,比化合物1分子质量多了34 Da,推断为M1结构中加2个羟基而形成。其二级质谱主要的特征碎片离子(见图13)有m/z207.066 2、167.070 3、105.034 0。其中m/z309.112 2碎片可能为M5中间链接羟基的碳链断裂形成,m/z167.070 3碎片可能为M5左侧苯环上的呋喃环打开形成,m/z105.033 5为M5右侧的苯环外连接一个羰基的结构,由上推测M5为M1加两个羟基形成。
2.4 药代动力学
按照经过方法学验证的UFLC-MS/MS方法测定大鼠尾静脉样品1给药后(10 mg/kg)的各个时间点的血药浓度,采用DAS 2.0进行计算药代动力学参数,并采用非房室模型对药物在大鼠体内的动力学过程进行拟合,尾静脉注射样品1后,测定大鼠血浆中的样品1及M1的浓度,药时曲线见图14。样品1的最大血药浓度Cmax=405.962 μg/L,达峰时间Tmax=0.083 h,半衰期T1/2=3.738 h,药时曲线下面积AUC0-t=190.635 g/(L·h),清除率Cl=65.578 kg·h/L,表观分布容积为Vd=299.378 L/kg,M1的最大血药浓度Cmax=281.291 g/L,达峰时间为Tmax=0.083 h,半衰期T1/2=3.011 h,药时曲线下面积AUC0-t=561.302 g/(L·h),清除率为Cl=19.179 kg·h/L,表观分布容积为Vd=79.032 L/kg,结果表明M1在5 min时就达到了最大浓度,样品1在体内转换成了代谢产物M1,而后M1浓度一直随样品1的减少而减少。

图9 M1可能的裂解机制Fig.9 Possible fragmentation pathways of M1

图10 M2可能的裂解机制Fig.10 Possible fragmentation pathways of M2
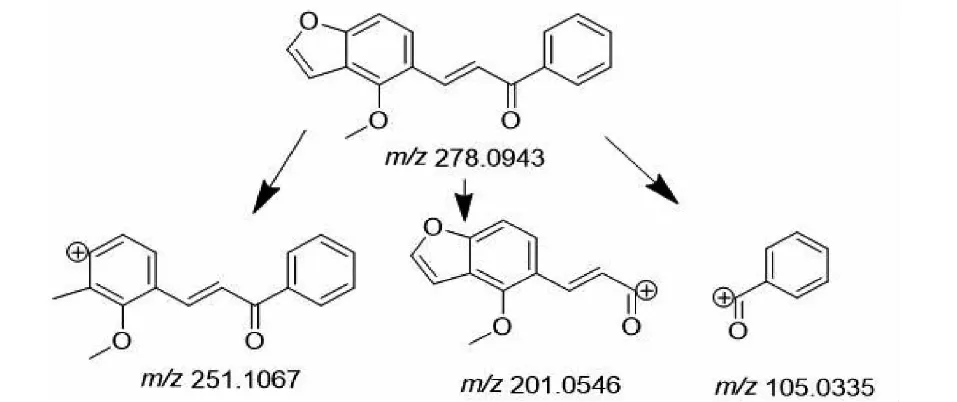
图11 M3可能的裂解机制Fig.11 Possible fragmentation pathways of M3
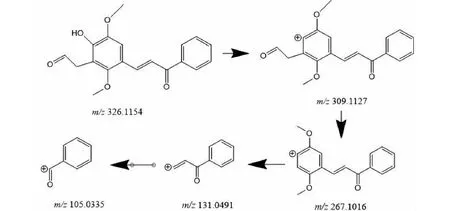
图12 M4可能的裂解机制Fig.12 Possible fragmentation pathways of M4
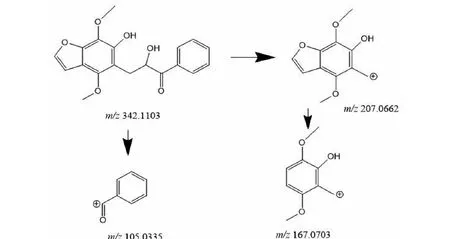
图13 M5可能的裂解机制Fig.13 Possible fragmentation pathways of M5

图14 样品1及M1在大鼠体内的药时曲线图(n=6)Fig.14 Concentration-time profiles of compound 1 and M1 in rats(n=6)
3 讨论与结论
绒毛崖豆藤是一味具有良好抗炎疗效的中药之一,样品1作为其主要的抗炎成分,国内外对其研究较少,尚未见其体内外代谢研究相关报道。本实验使用UPLC-QE-Orbitrap-MS方法具有较高的灵敏度及专属性,对于代谢产物的发现及鉴定具有显著的优势,同时鉴定出了样品1在小鼠、大鼠、猴、犬和人5个种属肝微粒体中的代谢产物为M1、M2,在体内有代谢产物M1、M2、M3、M4、M5,后根据质谱碎片推断出每个代谢产物的结构,并采用合成的方法制得M1,证明推断的化合物结构准确可靠。采用UFLC-MS/MS法对体内样本进行定量分析,通过方法学验证表明该方法稳定可靠,可以快速定量检查出样本中的微量目标化合物,相对于液相色谱法及紫外分析方法具有灵敏度更高等优势。
化合物的药代动力学参数是评价化合物的一个重要指标。通过体内外研究其主要代谢产物为M1,后在大鼠体内研究样品1及M1的代谢速率,并计算其药代动力学参数,发现样品1在体内转换为M1后再进行进一步代谢为M5。本研究对样品1进行了体内外代谢产物研究,推断出了5种代谢产物,并在大鼠体内测定了样品1及主要代谢产物M1的药代动力学参数,探究了样品1及M1在大鼠体内血浆中的变化趋势,结果表明化合物1在体内不稳定,快速代谢为M1,表明起抗炎作用的是其代谢产物而非自身,本研究为其阐明体内抗炎作用物质机制提供理论依据。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
现代仪器与医疗(2022年1期)2022-04-19
现代仪器与医疗(2021年6期)2022-01-18
粉末冶金技术(2021年3期)2021-07-28
昆明医科大学学报(2021年4期)2021-07-23
现代仪器与医疗(2021年2期)2021-07-21
康颐(2020年15期)2020-11-10
英语文摘(2020年7期)2020-09-21
分析化学(2019年3期)2019-03-30
分析化学(2018年12期)2018-01-22
