
表现为超长节段横贯性脊髓炎的视神经脊髓炎谱系疾病1例并文献复习
2022-07-28 08:52侯越王友明赵妍张如路
神经损伤与功能重建 2022年7期
侯越,王友明,赵妍,张如路
视神经脊髓炎谱系疾病(neuromyelitis optica spetrum disorders,NMOSD)是一种炎症性脱髓鞘疾病,以视神经和脊髓受累的中枢神经系统免疫病。其中以超长节段横贯性脊髓炎(ultra-longitudinally extensive transverse myelitis,u-LETM)或孤立性视神经炎(optic neuritis ON)起病,或反反复复且逐渐进展为NMOSD的一组高危综合征,短期内复发率高,预后较差[1]。但目前关于u-LETM 为主要临床经过的NMOSD的病例鲜有报道,本研究现将本院收治的1 例表现为u-LETM 的NMOSD 报道如下,以提高对本病的认识。
1 资料与方法
1.1 病例资料
患者,男,46岁,于2021年1月19日因“双下肢麻木1周,加重伴双下肢无力、排尿困难1 d”为主诉入院。患者于2021 年1 月12 日开始无明确诱因出现双下肢麻木,起初为左脚麻木,未予注意,麻木范围逐渐扩大,2 d 后出现右脚麻木,逐渐向上蔓延,前往当地医院就诊,并行胸椎MRI 示“脊髓长条状异常信号”,给予药物治疗,具体用药不详,病情仍持续进展,1月18日出现双下肢无力,左下肢为著,伴有尿潴留,无法排尿,给予膀胱造瘘,患者症状未见明显好转,为进一步治疗就诊于我院。病程期间无发热、头痛、头昏、肢体抽搐、恶心、呕吐、视力下降。既往体健。入院查体:神志清楚,言语流利,颅神经(-),双上肢肌力5级,双侧桡骨膜反射,肱二头肌、肱三头肌反射(++),左下肢肌力1+级,右下肢肌力2+级,双膝腱反射(-),双Babinski 征(+),提睾反射消失,双侧T8以下痛觉减退,脑膜刺激征阴性,心肺腹(-)。入院后辅助检查:血常规正常,尿常规:红细胞(++),脑脊液常规、生化未见异常,脑脊液抗水通道蛋白4(aquaporin 4,AQP4)抗体(+)1 ∶100、抗髓鞘少突胶质糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体(-)、抗胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)抗体(-)、抗髓鞘碱性蛋白(myelin basic protein,MBP)抗体阴性;血清AQP4抗体(+)1∶100;甲七项:甲状腺过氧化物酶抗体(thyroid peroxidase antibody,TPOAb)>300 IU/mL(正常0~30 IU/mL),甲状腺球蛋白抗体(thyroglobulin antibody,TGAb)>300 IU/mL(正常 0.21~30 IU/mL),TSH、FT3、FT4均正常;血沉26 mm/h(正常0~20 mm/h),ANA 弱阳性,C 反应蛋白、类风湿因子正常,HIV、梅毒(-),肿瘤5 项无异常;2021 年1 月21 日脊柱核磁平扫+增强示C6~T9椎管内脊髓异常信号,增强无明显强化,见图1。入院当天依据患者症状、体征及入院前检查初步诊断为“急性脊髓炎”,并给予甲泼尼龙500 mg/d 冲击治疗及B 族维生素等营养神经药物治疗,2021 年1 月20 日患者病情进展为双下肢截瘫,深浅感觉完全消失,考虑“超长节段横贯性脊髓炎”,并给予人免疫球蛋白27.5 g/d 静点后病情未再进展,结合患者症状、体征、脑脊液AQP4 阳性结果、影像学等表现,修正诊断为“视神经脊髓炎谱系疾病”,激素冲击治疗5 d 后改为口服60 mg/d 并逐渐减量,患者病情平稳,于2021年2 月5 日好转出院,出院后继续口服甲泼尼龙片56 mg/d,并逐渐减量维持治疗。出院后50 d 因突发胸闷、气喘入住我院急诊科,随即出现意识不清、呼吸困难等症状,家属拒绝行气管切开、呼吸机辅助呼吸等进一步治疗与检查,要求出院,予签字出院。
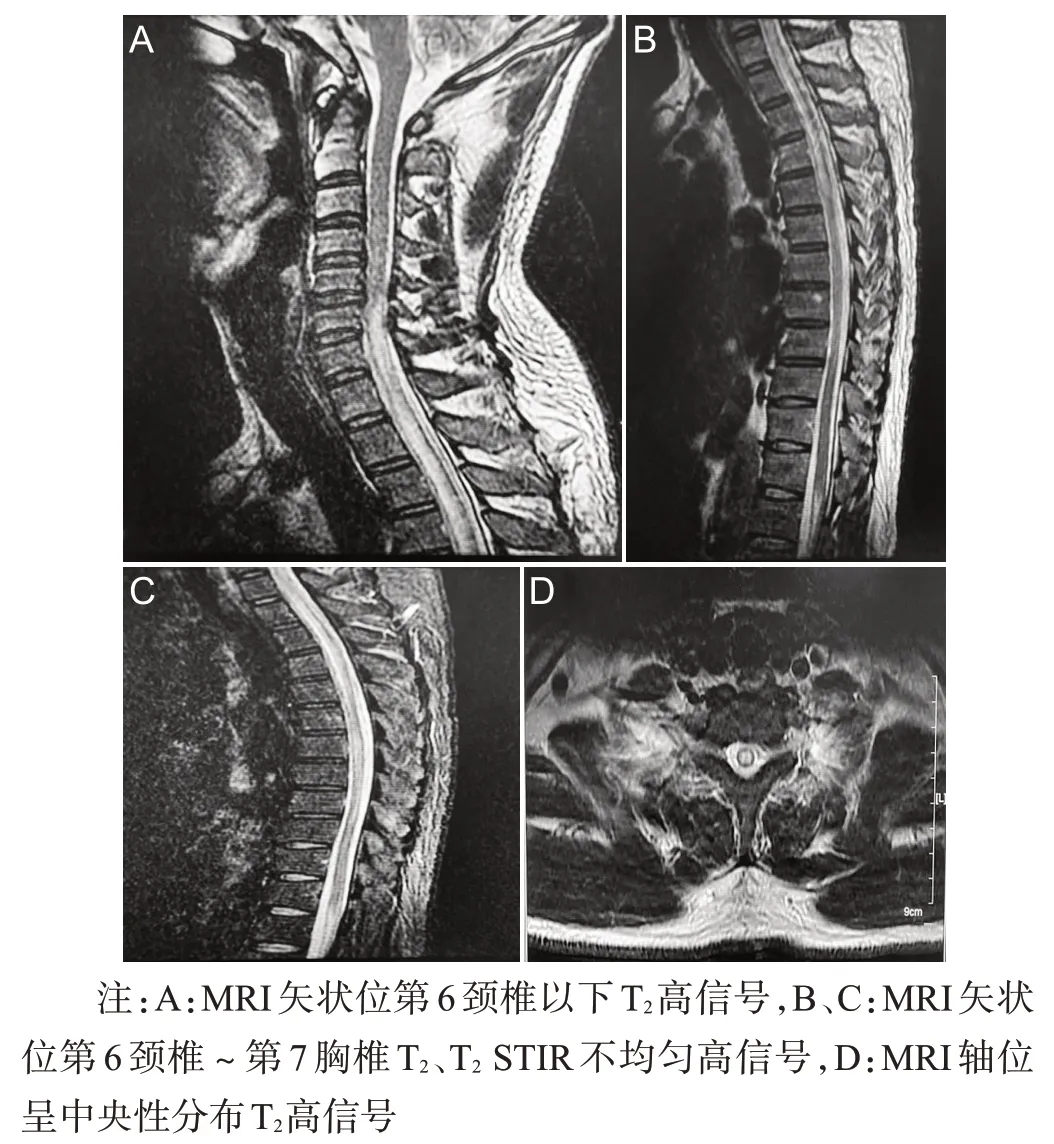
图1 颈胸椎MRI
1.2 方法
收集资料并分析。通过检索PubMed、谷歌学术、中国期刊全文数据库(CNKI)、万方数据库,收集关于表现为u-LETM 的NMOSD 的文献及病例报道:中文检索式:“超长节段横贯性脊髓炎”AND“视神经脊髓炎谱系疾病”;英文检索式“ultra-longitudinally extensive transverse myelitis”AND“neuromyelitis optica spetrum disorders”。检索时间自数据库建库至2021年4月。采用描述性统计对符合条件的所有病例从临床表现、实验室检查特点、急性期与缓解期治疗方案进行分析。
2 结果
本研究初步数据检索共获得相关文献25篇,经过剔除重复文献、阅读文题及摘要、全文筛选后,最终纳入8 篇文献。共检索到表现为u-LETM 的NMOSD 患者44 例。①临床表现:初始临床症状:横贯性脊髓炎(transverse myelitis,TM)24 例(54.5%)、视神经炎9 例(20.5%)、极后区综合征6 例(13.6%)、TM 合并极后区综合征3 例(6.8%)、大脑综合征2 例(4.5%),患者首次发病后复发率78%(32/41)(其中3例失访患者不纳入其中);②实验室检查:急性期AQP4抗体阳性率75%(33/44);其他自身免疫抗体阳性:抗SS 抗体17 例(38.6%)、抗甲状腺抗体(anti-thyroid antibodies,ATAbs)4 例(9.1%)、抗核抗体 9 例(20.4%),抗Ro-52抗体8例(18.2%);③治疗方案:急性期仅接受大剂量糖皮质激素冲击治疗30 例(68.2%),联合静脉注射人免疫球蛋白(intravenous immunoglobin,IVIg)7例(15.9%),联合环磷酰胺3例(6.8%),治疗性血浆置换4例(9.1%);缓解期口服小剂量激素19 例(43.2%),联合环磷酰胺5 例(11.4%),联合吗替麦考酚酯6例(13.6%),单用MMF8例(18.2%),单用硫唑嘌呤4例(9.1%)。
3 讨论
NMOSD 是一种危及生命、相对罕见的中枢神经系统自身免疫性疾病,临床为急性视神经炎、长节段横贯性脊髓炎(longitudinally extensive transverse myelitis,LETM)和延髓极后区表现,导致永久性神经功能缺损。AQP4 抗体是其特异性自身抗体,AQP4广泛分布于中枢神经系统,因此测定AQP4抗体可有助于早期诊断与治疗[2]。血清AQP4-Ab是u-LETM致病与复发的关键因素之一,检测血清AQP4-Ab可用于判断患者临床分期及疾病活动性[3]。
LETM 是一种相对罕见的脊髓综合征,Wingerchuk 等将其作为视神经脊髓炎(neuromyelitis optica,NMO)的一个关键支持标准和AQP4 抗体阳性的NMOSD 的主要类型[4,5]。然而,LETM 的大多数研究都集中在累及脊髓节段的下限,使用三个椎体的节点来区分NMOSD 和多发性硬化[6]。只有很少病例报告专门讨论较长的脊髓病变,而这些病例多是继发于系统性红斑狼疮和副肿瘤性脊髓病的LETM[7,8]。2011年张伟赫等[9]曾报告过4 例横断性脊髓炎伴全脊髓损伤(脊髓MRI 病灶长度>10个椎体节段),这些患者预后均较差,因此将这种更严重的横贯性脊髓炎称为u-LETM。
NMOSD 的主要治疗方法分为急性期治疗和缓解期治疗。目前对于急性期应用糖皮质激素冲击疗法仍是一线治疗方案,联合免疫球蛋白治疗可以减少NMOSD 的复发次数[11],缓解期使用药物多以硫唑嘌呤等免疫抑制剂或雷公藤多苷此类应用于自身免疫系统疾病的药物为主[12],目前CD19单克隆抗体此类针对NMOSD 的靶向药物已部分投入临床,有望推动NMOSD 患者的个体化治疗[13]。但有研究表明u-LETM患者在多次复发反复冲击治疗后有近40%患者出现脊髓萎缩[3],这仍是目前需要解决的难题。
然而并非所有的u-LETM 都由NMOSD 引起,一项关于u-LETM患者的回顾性研究表明超过三分之二的患者被诊断为NMOSD,而其中被诊断为NMOSD的患者中有三分之一的患者表现出与一种或多种系统性疾病的共病,但在非NMOSD 组中只有1 例患者表现出共病[10]。因此,系统性自身免疫疾病可能有助于NMOSD的诊断。
本例患者同样合并系统性自身免疫疾病,其血清ATAbs阳性,是自身免疫性甲状腺疾病(autoimmune thyroid disease,AITD)的特异性指标。由于甲状腺激素具有促进神经系统发育、增强神经系统兴奋性等作用,此外在中枢神经髓鞘形成和髓鞘破坏中也起重要作用,因此ATAbs 可与MBP形成免疫复合物诱导脱髓鞘[14]。有研究表明ANA和ATAbs有较密切的关系,多数ATAbs阳性患者常合并ANA阳性[15,16],而ANA 引起的炎性细胞因子基因表达的变化可导致血脑屏障破坏,或产生AQP4 诱导NMOSD 样作用所需的炎症环境[17]。ATAbs 阳性的NMOSD患者的病情更重,提示自身免疫性疾病患者的免疫状态比非自身免疫性疾病患者更活跃,因此影响病情的严重程度[18],因此监测ATAbs可能对判断NMOSD的预后具有一定的作用。
本例患者为中年男性,双下肢截瘫、深浅感觉消失,颈胸椎核磁见C7~T9超长节段异常信号,符合u-LETM特征,AQP4抗体(+),依据2015年国际NMO 诊断小组提出的新诊断标准[19,20],NMOSD 诊断明确。患者无明显甲状腺功能亢进或减退症状,但患者TPOAb、TGAb阳性,TSH、FT3、FT4均正常,甲状腺超声未见异常,结合内分泌科会诊意见,故排除桥本甲状腺炎(Hashimoto thyroiditis,HT)及Graves 病,出院后该患者短期便出现复发,病情进展迅速,预后差。
u-LETM是一种与NMOSD密切相关的病因异质性的脊髓综合征,且血清AQP4-IgG 阳性率与脊髓受累程度呈正相关。极后区综合征和大脑综合征此类表现是鉴别NMOSD与其他疾病的关键点。此外,女性多见、急性或亚急性起病、合并其他全身性免疫系统、颈胸病变等其他参数也有助于诊断NMOSD 相关的u-LETM。本例患者首次发病时未出现视神经炎、极后区综合征等表现,容易误诊和漏诊,详尽的病史及查体、AQP4 抗体的筛查,头颅及脊椎核磁有助于本病的诊断,表现为u-LETM的NMOSD的患者往往短期复发率高,预后较差,因此小剂量激素和免疫抑制剂的应用对于缓解期的治疗和改善预后而言尤为重要。