
肠道噬菌体组生物信息学分析方法的研究进展
2022-07-26 02:23胡晴玥李德志
微生物学杂志 2022年3期
胡晴玥, 李德志, 刘 箐
(上海理工大学 健康科学与工程学院,上海 200093)
病毒是地球上数量最多的生物实体,分布在生物圈的每一个角落,其中噬菌体是感染细菌的病毒,据估计在地球上的数量约为1031个,可能超过所有物种数量之和[1]。噬菌体广泛存在于身体的各个部位,如皮肤、口腔、肺部、肠道和尿道[2],其中肠道中噬菌体数量尤为庞大,研究估计健康成人的每克粪便约有109~1010数量级的噬菌体[3-4]。一方面,肠道噬菌体通过与宿主细菌的相互作用,调节肠道菌群的结构,如消灭宿主细菌,传递新的细菌表型,调节细菌群落组成以及基因表达和进化,进而影响动物宿主的生理健康[5];另一方面,噬菌体与哺乳动物细胞的病毒有一些共同特征,可被先天宿主受体(如Toll样受体家族)识别,调节免疫系统[6]。肠道噬菌体通过胞吞转运的方式跨过肠道上皮细胞进入动物宿主体内血液、淋巴、器官甚至大脑,直接与体内的细胞及器官相互作用,进而直接影响哺乳动物免疫系统和生理健康[7]。早期研究人员从肠道微生物群出发,探索并奠定了肠道微生物对人体健康及功能的重要作用,提出了健康的肠道微生物群(Healthy gut microbiome)这一概念[8]。而肠道噬菌体对哺乳动物的生理健康同样有着不可忽视的重要影响,因此有学者受到启发,从噬菌体的角度出发,提出健康的肠道噬菌体群(Healthy gut phageome),发现不同动物个体间噬菌体差异较大,但在健康个体间存在着一些共有噬菌体,组成健康肠道噬菌体组 (Phageome)。虽然肠道内噬菌体对人体健康以及疾病治疗有着重要的作用,但是对噬菌体的了解相对于其宿主细菌而言只是些零星的数据[9-10],主要原因:①传统实验室培养方法的局限;②噬菌体基因组的特殊特性;③相较于细菌而言缺乏通用的基因标记;④病毒组数据库非常不完善,超过80%的病毒缺少数据库信息,早期研究发现大多数已鉴定的肠道噬菌体与数据库中的序列没有同源性,不同研究之间变异性很高[10];⑤缺乏病毒分析标准。随着高通量测序和生物信息学以及机器学习技术的发展,许多以研究病毒组为目的的生物信息分析软件的开发与应用,使得研究人员可以深入探寻肠道噬菌体对人体的影响。Gregory等[11]整合来自32个研究项目的2 697个肠道宏基因组,含有33 242个病毒群(长度大于5 kb),覆盖来自16个国家的1 986名个体,构建了人类肠道病毒基因组数据库(Human Gut Virome Database,GVD)。为了进一步推进对肠道噬菌体多样性、进化分支以及全球分布情况的分析,Camarillo-Guerrero等[12]将来自6个大洲28个国家的28 060个人类肠道宏基因组,共142 809个非冗余噬菌体基因组和2 898个培养的肠道细菌基因组汇编入肠道噬菌体数据库(GPD),揭示了噬菌体的多样性以及宿主肠道细菌范围,类似病毒数据库的建立和逐步完善帮助原始数据在下游分析过程中进行分类和功能注释,提高了分析的准确性。通过生物信息学技术,可以揭示噬菌体的多样性、进化分支、相应宿主细菌以及功能,了解肠道噬菌体组在疾病中的角色,进而为疾病的诊断与治疗提供新的策略。肠道宏噬菌体组学的分析流程包括以下步骤:①原始数据质量控制和预处理,主要是过滤接头序列、低质量序列(quality scores<30),以及来自宿主动物或细菌等其他非病毒基因组序列;②肠道宏噬菌体组的拼接组装;③评估组装质量;④类病毒颗粒的筛选以及系统分类和功能注释;⑤进化分析和预测宿主细菌。对于肠道噬菌体组的相关研究学者,尤其是没有生物信息学背景的研究人员来说,选择合适的软件和分析方法成为一项挑战。本文将概述现阶段肠道噬菌体组数据分析主流的方法和思路,并对涉及到的软件工具和数据库进行详细介绍(图1)。
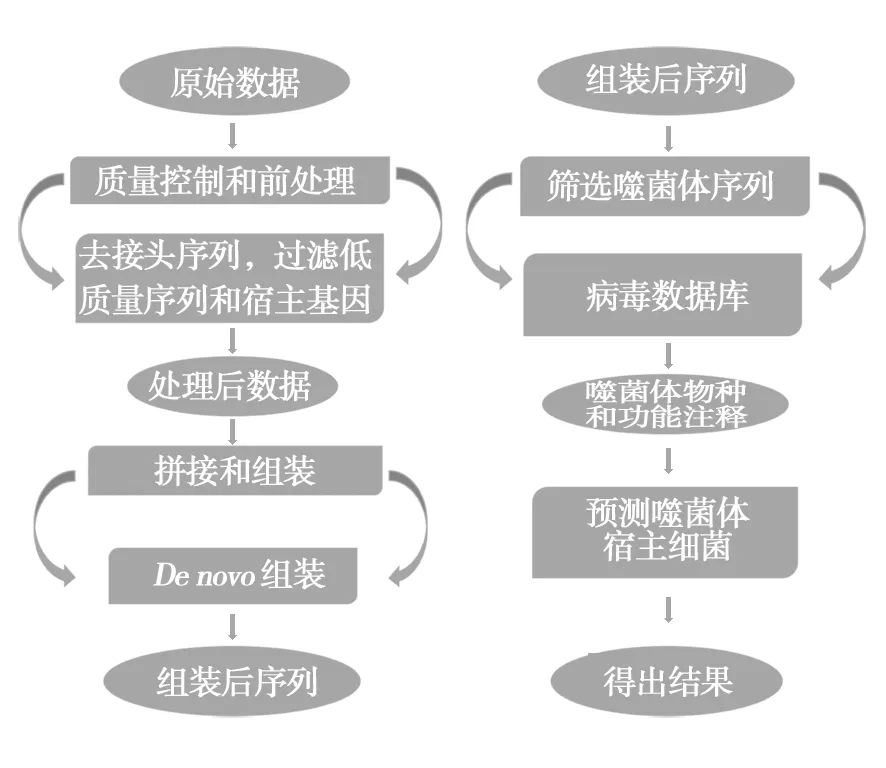
图1 肠道噬菌体组生物信息分析流程Fig.1 Bioinformatics analysis workflow of gut phageome
1 原始数据质量控制和预处理
样品测序后得到的原始数据(raw data)中包含许多非噬菌体基因组序列,例如构建文库时所产生的接头序列、引物序列和在提取噬菌体过程中残留的宿主基因等其他非病毒基因,以及由于测序错误而产生的低质量序列,非目标基因序列的存在会影响后续的下游分析,为避免分析结果受到影响,需要将这些序列去除。最常见的质量分析评估软件为FastQC,在Windows、Linux和Mac操作系统中均可使用,FastQC是基于Java所编译的,输出的结果以网页HTML的形式呈现。通过质量评估得出的结果对数据进行相应的预处理。Cutadapt[13]是常用的数据预处理软件,可以去除接头序列和超出指定范围长度的序列以及低质量序列(序列中含有较多的“N”),支持FASTA和FASTQ格式的文件输入和输出。在研究过程中常使用Minion[14]预测原始数据中的未知接头序列,其默认输入格式为FASTQ,需要将预测结果通过NCBI上的BLASTN与核酸数据库进行比对,确定其为生物学序列后再使用Cutadapt将其除去。AfterQC[15]是用Python开发的质控软件,可以发现并纠正绝大部分的错误序列,并具备高度自动化的数据过滤功能,还提供了校正重叠区域错误碱基以及预估序列错误率的功能,AfterQC可以自动检测和设置所有选项,大多数情况下的运行过程不需要添加参数和选项,方便研究人员使用。Trimmomatic[16]是适用于Illumina测序数据的质量控制过滤工具,常作为功能性软件嵌合在16S扩增子测序分析流程中[17],也可用于宏基因组数据前期的质量过滤和序列修剪。Fastp[18]是同时具备FastQC、Cutadapt、 Trimmomatic、AfterQC 四款软件中主要功能的数据前处理工具(表1),区别于前面提到的使用Java和Python编写的软件,Fastp使用C/C++语言汇编而成,可以实现多线程运行,运行速度比同类软件快2~5倍。
由于样本中含有一定比例的动物宿主基因,所以需要去除宿主基因或其他污染序列,再进行后续分析。通常使用比对参考基因组的方式识别其他来源的基因组,使用Bowtie2[19]、BWA[20]、BLAST[21]将数据与宿主参考基因组比对,识别出的污染序列可使用samtools[22-23]工具去除。FastQ Screen[24]同时包含质量控制和去除非病毒来源污染基因组的功能,可以同时比对多种不同来源的参考基因组,通过嵌套Bowtie1、Bowtie2、BWA三款序列比对软件包来识别其他污染序列,结果以文本和图形两种形式展现。MultiQC[25]可将所有样本的输出信息汇集到一个表格及图形文件中,便于研究人员比较样本间数据质量差异,在肠道噬菌体的分析中常用于对数据分析流程处理结果的评估。Aozan[26]通过嵌套FastQC、MultiQC、FastQ Screen三款工具,可自动处理原始数据(表2)。

表1 质量控制软件的数据输入输出格式和特点

表2 原始测序数据质量控制和数据前处理软件工具

续表2
2 噬菌体基因组序列的拼接组装
原始序列经过质量控制及预处理后得到的短序列需要拼接组装成较长的序列(contigs)才能进行后续的物种注释等下游分析。基因的组装算法主要分为Comparative组装和Denovo组装两类,Comparative组装是利用参考数据库中的同源序列来指导新基因组的构建;Denovo组装是将大量(短或长)DNA片段重新组装的方法,无需预先知道这些DNA片段的正确序列或顺序,在没有参考序列的情况下对未知基因组进行序列拼接[28]。由于目前数据库对自然界病毒种类的覆盖率不到20%,许多噬菌体在参考数据库中很难找到同源物,所以针对肠道噬菌体宏基因组的研究主要是通过Denovo组装来进行后续的下游分析。Denovo组装软件根据不同的策略主要分为三类:①基于Greedy策略的组装,这种方法在早期的基因组研究中比较常见,代表软件有TIGR[29]、VCAKE[30],此类组装策略的缺点是在合并reads或contigs的过程中过度注重局部序列最优化的选择而忽视序列之间的整体关系,从而导致在重复序列的组装中会出现错误;②基于Overlap-Layout-Consensus策略的组装,适用于较长reads的测序数据(Sanger、PacBio、Nanopore),常用的代表软件有Celera Assembler[31]、Canu[32]、Falcon[33]、Arachne[34]、MIRA[35]。Celera Assembler的出现极大推动了基因组学的研究,多细胞生物的第一个全基因组霰弹枪测序序列[36]和人类个体第一个二倍体序列[37]就是由Celera Assembler组装完成的。Canu是Celera Assembler的一个分支,适用于高噪声的单分子测序(PacBio RSII和Oxford Nanopore)。Falcon适用于复杂基因组的组装。MIRA和Arachne支持拼接组装全基因组霰弹枪序列;③基于DeBruijnGraph策略的组装,适用于序列长度较短的Illumina等二代测序数据,是分析肠道噬菌体组中最常用的一种组装策略,常用的软件有IDBA-UD[38]、SPAdes[39]、Megahit[40]、SOAPdenovo2[41]、SKESA[42]。IDBA-UD适用于组装测序深度不均匀的短读长序列,使用多个深度相对阈值来过滤低深度和高深度区域的错误k-mers,以及采用双端局部组装技术解决低深度短重复区域的分支问题,准确性要高于其他同类的短序列组装软件,但资源消耗较高。Megahit采用了简洁版的DeBruijngraph算法,在运行时间和内存需求方面相对于其他同类组装软件具有优势,适用于组装大型复杂的宏基因组数据,在大量样本混合组装方面优势明显,运行速度很快,对硬件设备资源的消耗少(需要运行内存约30 G)。SOAPdenovo常用来组装许多大型真核生物基因组,也可用于细菌和病毒基因组的组装,SOAPdenovo2在SOAPdenovo的基础上改进了纠错算法,减少DeBruijnGraph构造过程中的内存消耗,在contigs组装中能够解析较长的重复区域,并且组装得到的contigs数量较多,增加了组装长度和scaffolding的覆盖范围,适用于较大基因组的短序列组装,组装速度快,但是错误率较高。SPAdes是应用最广泛、各项指标参数最突出的组装工具,提供了很多样品类型的分析技术,当前版本适用于Illumina和IonTorrent测序数据,并且支持PacBio、Nanopore和Sanger测序数据的混合组装,SPAdes由八种不同的组装流程包构成,用于宏基因组和宏转录组的组装,可以从宏基因组数据集中组装质粒和生物合成基因簇,其中内嵌的metaSPAdes[43]包是目前宏基因组领域组装指标较好的软件,组装得到的contigs数较多,不足之处是拼接时间较长,运行过程设备的资源消耗较高,需要较大的运行内存(约250 G),适用于对肠道噬菌体组进行拼接。SKESA是近几年新开发的组装软件,适用于Illumina测序数据的组装,可以过滤污染序列,得到的contigs具有较高的质量和连续性,其运行速度相较于SPAdes、MEGAHIT有明显的提升,组装得到的N 50平均长度高于上述常用的两种拼接软件,并且组装错误率较低,现阶段主要用于拼接SRA数据库中的微生物基因组,并且嵌套于Pathogen Detection Project(PDP)分析流程中。序列组装完成后通常需要评估组装质量,常用的工具有QUAST[44]、MetaQUAST、CheckV。QUAST支持FASTA格式的contigs和参考序列以及FASTQ、SAM和BAM格式[45]的数据,QUAST融合了现有软件(Plantagora、 GAGE、 GeneMark.hmm[46]、GlimmerHMM[47])的质量度量方法,并对其进行了扩展,既可以通过比对参考基因组来评估已知物种的组装质量,也可以计算评估缺少参考基因组的未知物种,评估结果以图表的形式输出。MetaQUAST[48]是在QUAST基础上衍生出的更加先进的宏基因组组装评估软件,可以同时比对多个参考基因,并制作多个基因组的组装质量评估图表,在分析评估常见的未知物种时,会自动检测并从NCBI数据库中下载相近的同源参考序列以提高评估准确性,可以检测嵌合序列并报告“种间装配错误”。CheckV[49]可以自动评估宏病毒组组装的完整性和病毒组single-contig中的宿主污染情况,通常将CheckV与MetaQUAST结合使用以准确评估噬菌体组的组装质量(表3)。

表3 病毒基因组的拼接组装和评估序列组装质量工具
3 类病毒颗粒的筛选和系统分类注释
组装后的基因组需要确定其是否为噬菌体序列,因此需要注释和筛选出病毒基因组,对噬菌体组进行系统分类和注释是功能分析的关键步骤,也是研究肠道噬菌体的重要问题[50]。系统分类和注释的方法根据原理不同可分为两类,一类是基于将序列或组装得到的contigs与参考数据库进行比对,常用BLAST中的tBLASTx、BLASTn、BLASTx[51]工具对序列进行比对注释,或将组装得到的scaffolds与NCBI中的Refseq virus数据库进行比对(ftp://ftp.ncbi.nlm.nih.gov/refseq/release/viral)。国际病毒分类委员会(ICTV)存储了病毒分类、分类单元名称和相关的宏病毒组数据,包括每个已命名物种的示例病毒信息,ICTV官方网站(https://talk.ictvonline.org/)内含病毒分类数据库,对每种经过定义的病毒链进行了完整的描述[52]。pVOGs[53](Prokaryotic Virus Orthologous Groups)数据库包含近3 000个完整的原核宿主病毒基因组(超过97%为噬菌体)和9 518个直系同源组,该数据库可以应用于分析已知噬菌体的进化分类、了解病毒蛋白家族的历史、噬菌体基因组的重建以及帮助特征不佳的基因组注释同源基因。IMG/VR[54](The Integrated Microbial Genome/Virus)是目前最大的致力于研究病毒组学的公共数据管理和分析平台,最新版本的IMG/VR包含18 373个已培养和2 314 329个未培养的病毒基因组,可以进行基因组的注释以及预测宿主细菌分类,支持用户根据基因组特征或序列相似性高效地浏览、搜索和选择未培养的病毒基因组。PHASTER[55]是用于对细菌和质粒中的前噬菌体进行快速鉴定和注释的网页服务器,输入文件支持FASTA格式的原始核酸序列或GenBank格式的已注释基因组数据,也可以通过数据库中的序列编号对相应参考序列进行分析,分析结果以图表的形式直观展现,PHASTER提供用户友好型的图形交互界面便于研究人员使用,不足之处是单次上传的文件大小不能超过40 M,需要使用脚本将FASTA文件分为多个小文件,并且网页服务器运行不稳定。
由于噬菌体基因组系统发育受到类群内广泛的水平基因转移和基因组模块化的损害,导致环境样本中大量的噬菌体颗粒复杂化增加,使得在参考数据库中查找噬菌体的同源序列变得非常困难[56],并且相关数据库包含的信息有限,采用同源序列比对的方法会有大量序列被标记为“未知”[57]。为解决这些难题,许多分析工具使用机器学习或深度学习的算法来进行病毒的系统分类和注释,有效解决了基于参考数据库和同源序列比对的方法不能从宏基因组数据中识别未知病毒或短病毒序列的问题。Prophage Hunter[58]提供一站式的网络服务,从细菌中提取原噬菌体基因组并评估其活性,识别系统遗传学相关的噬菌体,并注释噬菌体蛋白功能,Prophage Hunter在建库的过程中采用了基于序列相似性的搜索和噬菌体遗传特征的机器学习分类算法,能够识别参考数据库之外的未知噬菌体。更多的宏基因组功能注释方法可参考Prakash等[50]的文章。MARVEL[59]采用随机森林机器学习的方法来预测筛选宏基因组数据中的双链DNA噬菌体,筛选结果具有较高的准确性。RNN-VirSeeker[60]是基于长短期记忆网络模型构建的病毒识别软件,模型通过数据库进行训练,软件可以自动查询基因组的高级特征,并根据softmax层的评分来预测病毒序列,在识别筛选较短的病毒序列(<500 bp)和人体肠道宏噬菌体组时具有较高的准确性。VirSorter[61]是基于隐马尔科夫模型(Hidden Markov model,HMM)建立的,能够以较高的准确性检测多种类型微生物的较短(3 kb)组装序列(contigs)中的病毒信号,使用RefSeqABVir或Viromes[62]作为参考数据库。VIBRANT[63]使用混合机器学习和蛋白质相似性方法,从宏基因组组装序列中表征病毒群落功能,突出病毒辅助代谢基因和代谢途径,利用蛋白质特征的神经网络和新开发的v-score度量标准,可以最大限度地识别裂解噬菌体基因组,包括高度多样化的噬菌体组。病毒和宿主有明显不同的k-mer特征,通过基因组k-mer频率分布来发掘病毒基因是常用的筛选肠道噬菌体组的分类鉴定方法,这类方法的代表软件是VirFinder[64],它是第一个使用k-mer频率的机器学习方法来筛选病毒序列。ViraMiner[65]包含了卷积神经网络(Convolutional Neural Networks,CNN)的两个分支,用于检测原始宏基因组contigs中病毒模式和模式频率,可以从不同人类样本的原始宏基因组序列中检测噬菌体序列。DeepVirFinder[66]同样采用卷积神经网络的深度学习方法来自动学习病毒基因组特征,并同时基于这些基因组特征建立预测模型来判断序列是否源自噬菌体基因组,在研究分析中,通常将MARVEL与DeepVirFinder组合使用的准确性和覆盖率较高(表4)。

表4 类病毒基因组的筛选、系统分类注释软件以及常用的病毒数据库
4 进化分析和预测相应宿主细菌
肠道噬菌体对相应宿主细菌群落的调节和生化作用会直接或间接的影响哺乳动物生理健康,了解噬菌体-宿主的感染性,对于从微生物群系出发理解噬菌体对细胞生命的影响以及它们作为肠道生态系统的重要组成部分至关重要。研究人员发现整合到宿主基因组的溶原噬菌体拥有与宿主细菌tRNA基因完全匹配的附着位点[67],因此Bellas等[68]将噬菌体基因组scaffolds通过BLASTN比对到tRNADB-CE数据库中,用匹配tRNA的方法在门或纲的水平上推测宿主细菌。Boeckaerts等[69]通过构建受体结合蛋白(RBP)序列数据库,使用机器学习的方法在种水平上预测噬菌体的宿主细菌。Young等[70]结合噬菌体基因组的k-mer组成和蛋白结构域,开发了一种新的预测宿主细菌的计算框架,通过研究噬菌体基因组的核苷酸、氨基酸特性和蛋白质结构域等特征,并结合这些互补特征,提高宿主预测的准确性,认为感染同一宿主的噬菌体之间进化关系所产生的系统发育信号也可以被预测,这是由于在共同进化过程中尽管发生了频繁的宿主切换,但病毒和宿主系统发育树倾向于一致。Villarroel等[71]开发了HostPhinder工具,该工具通过比较k-mers,根据基因组相似性预测噬菌体的细菌宿主,HostPhinder支持交互式网站服务。Ahlgren等[72]利用基于病毒和宿主寡核苷酸频率模式来预测给定病毒的宿主细菌,并且提供了VirHostMatcher程序用于计算寡核苷酸频率(ONF)分数以及结果的可视化。Galiez等[73]开发的WIsH软件使用齐次马尔可夫模型(Homogeneous Markov Model)预测噬菌体contigs中的细菌宿主,WIsH可以快速准确的预测短噬菌体序列的宿主。Leite等[74-75]基于噬菌体和宿主细菌结构域相互作用的得分情况和蛋白质一级结构信息,应用机器学习的方法预测相应宿主细菌。Lu等[76]推出了原核病毒宿主预测器(Prokaryotic Virus Host Predictor,PHP),利用病毒和宿主基因组序列之间的k-mer频率差异作为特征来预测原核病毒,PHP是使用高斯模型构建的交互式网页服务器,输入文件支持FASTA格式的纯核酸序列。相关机器学习的方法在识别噬菌体宿主细菌方面的应用研究请参考Nami等[77]的文章。也有学者通过单细胞病毒荧光标记的实验方法来预测和鉴别人类肠道中噬菌体相应的宿主细菌[78],而生物信息软件利用噬菌体和宿主细菌之间共有的生物特征或生物大分子模式通过机器学习的方法来预测宿主细菌,相较于实验方法可以在短时间内大批量分析鉴别多个样本中的噬菌体宿主细菌,提高研究效率,避免实验操作中出现的误差。
系统发育分析用于研究基因或物种之间的历史关系,并以分支图的形式描述这些关系,称为系统发育树(Phylogenetic analysis)。构建系统发育树常用的软件有MEGA[79]、ggtree[80]、FastTree[81]、Cytoscape[82],数理统计分析及可视化展现通常使用R语言来完成,常用的R包有ggplot2、ggplot、ggiraph、ggfortify,有关使用R语言进行数据分析的详细内容可参考Chan[83]的文章。IMP[84](Integrated Meta-Omics Pipeline)是基于Denovo组装的开源生物信息分析流程,可对多组学数据集进行标准化、自动化、可重复的大规模集成分析,IMP嵌套了多种生物信息分析工具用于实现流程一体化运行,包含的功能有原始数据的预处理、宏基因组或宏转录组数据的迭代共组装、微生物群落结构和功能的分析、自动装箱分类以及基于基因组特征的可视化展现(表5)。

表5 预测相应宿主细菌和构建系统发育树的软件工具
5 展 望
肠道内存在大量噬菌体,它们在调节肠道微生物群落动态平衡,动物宿主的生理机能与免疫系统中发挥着至关重要的作用。随着高通量测序技术的发展以及相应生物信息分析软件的开发与应用,使许多肠道病毒基因组数据可以被获取,相关数据库可以覆盖种类更加丰富的病毒基因组,如GPD、ICTV、pVOGs、MG/VR、Viromes等数据库的出现,让人们对肠道噬菌体以及其对肠道菌群和宿主生理健康的影响有了更广泛的认识。本文主要对肠道噬菌体宏基因组的分析流程和所需的相关工具软件以及数据库进行综述。机器学习和数理统计算法的兴起,使得肠道噬菌体组的分析不再仅局限于使用参考数据库和序列比对的方法,但是对噬菌体功能注释和筛选的准确性要低于序列比对得出的结果。并且宏基因组的数据量一般较大,在一些分析步骤中(如序列拼接组装和序列比对)会占用较大的内存和主存(RAM)空间,硬件配置较低的计算机在运行过程中会因此报错,且运行时间较长。病毒基因变异性强,只依据机器学习算法筛选噬菌体基因组或对其进行功能分析会有较大的误差。因为许多肠道细菌不能通过传统的体外培养技术存活,所以这些肠道细菌对应的噬菌体无法有效分离纯化,进而造成运用传统的分离-纯化-测序的研究思路分析噬菌体存在一定的局限性。肠道噬菌体组学另辟蹊径,避开分离纯化这个步骤,直接分析肠道噬菌体的结构和组成,具有一定的先进性,但是需要将生物信息分析与实验观察分析相结合,以提高结果的准确性。目前大多数噬菌体的功能特性仍然无法得到解释,测序方法得到的噬菌体组序列大部分是新序列,噬菌体组的分类和功能注释依赖于病毒序列数据库,而数据库中记录的病毒信息是通过实验培养,质谱分析和分子生物学研究等得出的结论,其宿主范围,详细的生物功能和形态记录只能通过实验培养噬菌体来明确[86],体外培养噬菌体技术的限制导致现有的数据库无法对许多新噬菌体组序列进行分类和功能注释,这也是噬菌体组研究目前面临的瓶颈,并且需要开发通过更加精简的算法和脚本构建而成的生物信息分析工具,以减少软件在运行过程中对硬件的消耗。
猜你喜欢
军事文摘(2022年16期)2022-08-24
昆明医科大学学报(2022年2期)2022-03-29
植物保护(2021年4期)2021-11-12
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
当代水产(2020年3期)2020-06-15
科学24小时(2020年4期)2020-05-14
小星星·阅读100分(高年级)(2015年11期)2015-11-28
大自然探索(2015年11期)2015-09-10
