
不同茶树品种土壤细菌群落结构和多样性特征
2022-07-26 01:00:18黎巷汝陈丹妮洪永聪
茶叶学报 2022年2期
黎巷汝,陈丹妮,洪永聪*
(1.福建农林大学园艺学院,福建 福州 350002;2.武夷学院茶与食品学院,福建 武夷山 354300)
茶树是我国广泛种植的经济作物之一,2020年其种植面积达到316.51万公顷[1]。茶叶的产量和品质与品种特性、种植管理紧密联系,如何有效利用土壤养分,提高茶树的养分吸收效率是我国茶叶种植上亟待解决的问题[2]。植物主要通过根部吸收养分来满足自身生长生理需求,其根系-土壤-微生物形成了复杂的生态调控网络。土壤和植物的健康状况以及土壤养分库的丰富程度与土壤微生物群落的结构和功能密切相关[3-7],因此了解我国茶树根际微生物群落的生态特征茶园的养分管理有一定指导意义。
目前,已有诸多研究报道了茶树根际细菌群落的种类和作用。茶树根际存在多种有益菌,如:圆褐固氮菌(Azotobacter chroococcum)、环状芽孢杆菌(Bacillus circulans)、固氮梭菌等具有固氮作用[8];纤维素分解细菌、氨化细菌、芽孢杆菌(Baciluss)、假单胞菌(Pseudomonsadaceae)利于有机态养分的转化,提升土壤肥力[9-10];树根际中特定微生物菌株也具有提高茶树的耐胁迫性[11]及防治根腐病的能力[7],从而促进茶树生长和提高生产力。此外,不同技术手段获得的茶树根际微生物菌群有所差异。利用培养法[12-13]获得的优势菌主要为芽孢杆菌(Baciluss)和假单孢菌(Pseudomonas)。而采用高通量测序技术[14]研究发现茶树根际的优势细菌类群为变形菌门(Proteobacteria)、拟杆菌(Bacteroidetes)、酸杆菌(Acidobacteria)。这些研究揭示了茶树群落的共有特征,但目前有关不同茶树品种间的微生物群落特征研究却较少。其中多数研究[15-16]基于平板培养的方法寻找不同品种的群落差异性,不同茶树品种间微生物群落存在差异,优良的茶树品种具有更高的群落多样性和丰度,且有益菌群更多。然而土壤中存在大量不可培养的微生物菌落,需要用新一代技术才能深入了解茶树的土壤微生物全貌。随着测序技术的发展,Illumina高通量测序技术已被广泛运用于土壤微生物组的研究中[17],通过扩增细菌的16S rRNA可明晰土壤样本中的绝大部分菌落。此技术在茶树根际微生物领域中的研究主要集中在树龄[18]、施肥状况[19]和栽培措施[20],对于不同品种茶树根际微生物的影响鲜有报道。顾松松[21]利用16S rRNA技术测试了5种茶树的根际微生物,发现不同茶树品种土壤微生物多样性和群落结构存在差异。
本研究对同一种植条件的地块中10种茶树品种的土壤微生物群落进行16S rRNA高通量测序测序,旨在探讨不同茶树品种土壤微生物群落特征,为茶园管理提供理论基础。
1 材料和方法
1.1 样品采集
样品采集于福建省武夷学院的茶树种质资源圃(117°99′E,27°73′N),该资源圃于2015年建成,为平地茶园。每一行栽种1个茶树品种,每行长度为20~30 m,大行距2 m,双行单株种植。所有茶树为同一时间移栽,树龄7年。茶园管理措施一致,于每年10月下旬施复合肥,3、6、9月进行人工除草。
2021年5月,在该种质资源圃内,选取10个茶树品种:尖波黄(JBH)、白鸡冠(BJG)、大红袍(DHP)、蜀永1号(SY1)、白叶1号(BY1)、中茶302(ZC302)、政和大白茶(ZHDBC)、福安大白茶(FADBC)、佛手(FS)和毛蟹(MX)为调查对象,其生物学特性见表1。分别采集10个茶树品种的根区土壤。具体方法:种植行内随机选取5株茶树,去除土壤表面凋落物及杂物,近根处采集深度为0~20 cm的土壤样本。每5株茶树根区土壤混合为一个样品,每个茶树品种土壤样品进行3次生物学重复。每个样品用无菌自封袋封装,放入冰盒运送回实验室,-80℃冰箱储存,直至检测。

表1 10个茶树品种的生物学特性Table 1 Biological characteristics of 10 tea cultivars

(接表1)
1.2 DNA提取和Illumina测序
使用FastDNA Spin试剂盒(MP Biomedicals,Solon,USA)提取土壤样品中的总DNA,在1%琼脂糖凝胶上检测DNA的完整性,使用NanoDrop 2000紫外分光光度计(Thermo Scientific,MA,USA)测量DNA质量和浓度。采用Illumina测序技术,测序由凌恩生物科技有限公司完成,使用引物为341F(5’-CCTAYGGGRBGCASCAG-3’) 和 806R(5’-GGACTACHVGGGTWTCAAT-3’),扩增 16S rRNA的V3-V4区。
1.3 数据处理和序列分析
QIIME 软件包分析原始的 Illumina HiSeq测序数据,去除低20的质量分数和低于300 bp的序列长度,获得有效序列。并生成OTU表,从该表获得Resample OTU表以标准化总序列。使用Usearch的唯一序列来定义操作分类单元(OTUs),将相似性为97%的序列分类为同一OTU。使用SILVA数据库和QIIME中实现的RDP分类器进行物种分类注释。基于测序深度最低数据量对OTU表进行抽平,利用R和Mothur程序进行微生物种群多样性分析。使用EXCEL进行数据统计,R和R中的Vegan、ggplot2程序包进行统计分析和图表绘制。
2 结果与分析
2.1 测序和质量控制
对10个茶树品种根际土壤样品进行高通量测序,共获得512504条有效序列和3566个OTU分类单元。从每个样本中抽取一定数量OTU序列绘制稀疏曲线(图1A)和物种累计曲线(图1B),曲线趋于平缓,表明测序深度和抽样量足够,足以反应样品中绝大部分物种的信息,可进行下一步分析。

图1 10个茶树品种土壤细菌测序的稀释曲线(A)和物种累计曲线(B)Fig. 1 Bacterial sequencing dilution curve (A) and species accumulation curve (B) on soils planted with 10 tea cultivars
2.2 土壤样品的细菌丰度和多样性
在R中使用Vegan包对OTUs表进行抽平,每个样本抽取至34350条优化序列,进行α-多样性分析。Chao、Shannon指数反应了细菌群落的丰度和多样性,结果如表2所示。Chao指数代表微生物群落的丰富度,指数越高群落则物种越丰富。Shannon指数反映群落多样性,指数越高则群落多样性越多。10个品种的物种丰度和群落多样性有明显差异,其中JBH的Chao和Shannon指数均为最高,为2644和6.39,具有最高的物种丰度和群落多样性。而ZC302的物种丰度和多样性均为最低,为 1964.59和 5.73。

表2 10个茶树品种根际土壤样品测序结果各分类单元数量及Alpha多样性分析Table 2 Quantity and alpha diversity of bacteria in rhizosphere soils planted with 10 different tea cultivars
2.3 不同茶树品种土壤细菌群落的组成结构
为进一步了解不同品种的细菌群落组成,根据OTU表的注释结果,在门、属水平上进行分析。在门分类上(图2A),10个茶树品种的优势微生物种群结构相似,丰度前11位分别为变形菌门(Proteobacteria)、酸杆菌门(Acidobacteriota)、绿弯菌门(Chloroflexi)、放线菌门(Actinobacteriota)、浮霉菌门(Planctomycetota)、拟杆菌门(Bacteroidota)、wps-2门、蓝细菌门(Cyanobacteria)、芽单胞菌门(Gemmatimonadota)、粘球菌门(Myxococcota)、Armatimonadota门。10个茶树品种的组成差异表现在丰富度上,其中ZC302的绿弯菌门(Chloroflexi)的丰度相比最大(30.67%);芽单胞菌门(Gemmatimonadota)的丰度最少(0.14%)。属分类水平上,各品种群落组成相似(图2B)。

图2 10个茶树品种细菌群落在不同分类(A. 门分类水平;B. 属分类水平)上的相对丰度Fig. 2 Relative abundance of bacterial communities at phylum level (A) and genus level (B) in soils planted with 10 tea cultivars
在有注释的优势菌属中,丰度排名前的5个菌属分别为酸杆菌属(Acidobacteriales)、颗粒菌(Granulicella)、罗河杆菌属(Rhodanobacter)、节杆菌属(Arthrobacter)、慢生根瘤菌属(Bradyrhizobium)。其中酸杆菌属是JBH、MX、SY1、BY1的主要优势菌属,ZHDBC的优势菌属为罗河杆菌属(10.04%),FS的优势菌属为JG30-KF-AS9(7.51%)。
2.4 不同茶树品种土壤细菌群落组成的相似性和差异性
为综合比较不同茶树品种间的组成相似性和差异性,采用花瓣图对特有OTU和共有OTU进行分析(图3)。不同品种的共有和特有物种占各自群落的比例不同,10个品种共有物种为582个,其中ZC302共有OTU比例最多(37.45%),DHP、BJG、BY1、ZHDBC、FADBC、SY1、JBH、MX、FS分别占29.74%、28.20%、28.91%、32.14%、27.86%、29.83%、26.81%、34.77%、30.83%。茶树的共有核心菌群为变形菌门、酸杆菌门、绿弯菌门、放线菌门。进一步挖掘不同品种间的特有菌群,对各品种的特有OTU进行属水平上的分析。在有注释的属类中,DHP的优势特有菌属为Microcoleus SAG 1449-1a和Cylindrospermum PCC-7417,JBH的特有菌属为Candidatus Solibacter。

图3 10个茶树品种OTUs花瓣图分析Fig. 3 Analysis of petal map of OTUs of 10 tea cultivars
为研究不同茶树品种细菌群落的结构差异,基于属分类水平(丰度前30)的Bray-Crutis距离,使用类平均法(average)对10个品种的物种组成进行层级聚类分析(图4A)。结果发现,BY1和SY1、ZHDBC和FADBC、DHP和BJG分别聚为一支,细菌群落组成最相似。ZC302单独一支,与其他9个品种差异较大,9个品种归为一个类群。酸杆菌属(Acidobacteriales)主要分布于SY1、BY1、JBH、BJG、MX、DHP中。NMDS分析结果显示(图4B),stress<0.2,其模型较可靠地能反映样本排序的真实分布。10个茶树品种的NMDS样本排序分布结果与聚类分析结果相应,ZC302与其他样本距离较远,仍差距较大。
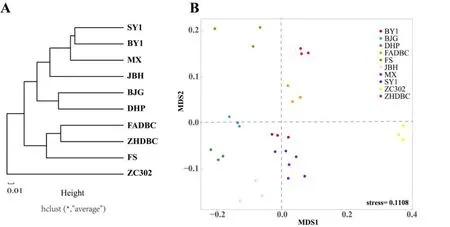
图4 基于属水平分类不同品种根际细菌的层级聚类分析和NMDS分析Fig. 4 Composition and clustering of dominant bacteria at genus level in soils planted with 10 tea cultivars
3 结论
本研究在同一地块、管理相对一致的种质资源圃中采集了10个不同品种茶树的根区土壤进行Illumina高通量测序,对不同茶树品种的土壤细菌群落多样性进行研究。α-多样性指数显示,10个茶树品种的多样性有所差异,JBH的菌群具有最高的多样性,ZC302的多样性最低。β-多样性分析10个品种间的群落相似性和差异,基于Bray-Crutis距离的层级聚类分析和NMDS分析都显示,10个品种根据菌属丰度可分为两个类群,ZC302与其他品种距离最远,差异较大;BY1、SY1和MX相似度最高。根际分泌物显著影响跟土壤微生物的种类和数量,而植物基因型对根际分泌物的产生有重要影响[22]。同一生境下,遗传背景相距较大的ZC302的细菌群落与其他品种差异较大。通过遗传背景调查发现,ZC302是以‘格鲁吉亚6号’为母本、‘福鼎大白茶’为父本育成的品种,而其他9个均为在我国本土生长繁育的品种。ZC302的母系来源于‘格鲁吉亚6号’,这可能是造成其根系土壤细菌群落与其他9个品种差异大的主要原因。在另外一个类群中,DHP和BJG、ZHDBC和FADBC群落相似度最高。DHP和BJG均为武夷山本地品种,ZHDBC和FADBC为均生长于福建北部,起源地相近或相同的两个品种具有相似的微生物群落结构,这可能与植物的环境适应相关[22]。
本研究中10个品种的细菌群落组成结构相似,其差异主要表现在群落丰度。在较大分类上,茶树的优势核心菌为变形菌门、酸杆菌门、绿弯菌门、放线菌门等,变形菌门和醋杆菌门占幅较大,与前人研究相似[14-21]。变形菌门、放线菌门、酸杆菌门是植物微生物群落的主要组成部分,只在不同物种中的表现不同丰度。变形菌门中包含了许多与土壤养分转化、光能营养相关的细菌种类。在所研究的茶树品种中,该门类下的罗河杆菌(Rhodanobacter)、慢生根瘤菌(Bradyrhizobium)、醋杆菌(Acidibacter)占据优势(图2B),其中罗河杆菌是反硝化菌菌群,慢生根瘤菌具有固氮作用,这两种菌均与营养转化和吸收相关[23-24]。在所有品种中酸杆菌属(Acidobacteriales)和颗粒菌属(Granulicella)占很大比例,两者均与植物凋零物的碳降解相关[25-26],说明茶树菌群功能偏向营养吸收和转化方面。各品种的特有菌属占各自总菌群的比例极低,且多为稀有菌群。
在同一生境下,来自不同地区的茶树品种根际细菌群落呈现出一定差异,地理起源及特征相近的品种群落多样性和丰度相似,茶树品种对根际细菌的组成结构和丰度有一定影响,但造成这些差异的原因还需要结合根系代谢组及基因组等其他信息进一步深入研究。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29 00:51:58
食品安全导刊(2021年20期)2021-08-30 06:40:50
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
乡村地理(2018年2期)2018-09-19 06:44:00
湖南农业(2016年3期)2016-06-05 09:37:36
音乐天地(音乐创作版)(2016年11期)2016-02-05 01:10:59
水生生物学报(2015年1期)2015-02-28 16:01:05
