
基于WGCNA分析狼疮肾炎的关键基因①
2022-07-21 09:15周豪坤丁兴红陈俊利黄惠莲钱俊华浙江中医药大学杭州310053
中国免疫学杂志 2022年10期
周豪坤 丁兴红 陈俊利 黄惠莲 钱俊华 (浙江中医药大学,杭州 310053)
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种因免疫系统对自身抗原免疫耐受减弱或消失而导致的自身免疫性疾病[1]。狼疮肾炎(lupus nephritis,LN)是SLE 的一种严重并发症,多以免疫细胞和细胞因子浸润、肾小球和肾小管间质损失为特征[2]。10%的患者在发病后5 年内会发展为终末期肾病(end stage renal disease,ESRD),需要通过透析或肾脏移植来维持生命[3]。LN 具有复杂的病理过程和较长的病程。目前诊断和治疗LN 的标准并不令人满意,而且不可能准确地预测患者对治疗的反应或个别患者的长期结果[4]。近年来,也许是因为更全面细致的狼疮管理方法,欧洲LN 患者的严重程度有所下降,但在北美及我国由LN 导致的ESRD发生率一直居高不下[5]。
免疫介导的炎症反应是LN 的主要启动因素[3],但其导致ESRD 的具体机制却不甚明确,目前还没有针对LN的生物疗法。现有的有关LN临床试验的设计中亦存在许多未解决的问题,从而影响了对试验结果的解释。因此迫切需要更有效的诊断方法,识别新的生物标志物和分子机制,以便早期发现和处理LN。
由于有效的诊断实验数量有限,一些研究通过高通量测序和生物信息学来研究与LN 相关的基因和/或分子特征,为识别LN 相关的关键基因、网络和通路提供了策略[6-7]。基因表达谱作为同时检测数千个基因的高通量数据可对其进行分析,以筛选出与疾病分子机制相关的显著差异表达基因(differen⁃tially expressed gene,DEG)。既往的高通量测序及基因芯片研究表明DEG 可通过不同信号通路、生物过程或分子机制参与LN 的发生发展[8-9]。然而,其下游分析鲜有涉及基因间的相关性以及基因与临床特征间的关系。加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)广泛应用于描述微阵列或RNA-seq中基因表达和临床特征之间复杂的相关性,并为疾病的诊断和治疗寻找潜在的生物标志物。其通过构建基因与表型间的基因共表达网络,发现与临床性状高度相关的重要模块,并筛选出枢纽基因,从而对探索疾病的分子机制提供思路[10]。与基因芯片和高通量测序分析相比,WGCNA 适合进行多种统计学检验,通过分析基因之间的相关性,避免根据某固定阈值筛选而丢失基因的变化趋势信息。
本研利用WGCNA 筛选与LN 高度相关的基因模块。根据关键模块和DEGs 的网络拓扑综合分析,对候选基因进行筛选,以发现新的候选生物标志物和治疗靶点。
1 材料与方法
1.1 数据的采集与预处理 本研究通过下载GEO数据库数据集GSE86425(包含54 例LN 小鼠肾脏组织样本和41 例正常小鼠组织样本)。数据集基于GPL11180 Affymetrix HT MG-430 PM Array Plate 微阵平台测序。应用R 语言对原始数据进行消除背景、标准化数据、基因注释、构建表达矩阵等预处理。去除缺失、重复及低表达的基因探针。
1.2 加权共表达网络构建 利用R 语言WGCNA包[10]构建加权共表达网络。使用pickSoftThreshold函数获得相邻函数加权参数的最优值,将其作为软阈值,用于后续网络构建[11]。然后构建加权邻接矩阵,并基于拓扑重叠矩阵(TOM)的相异度度量(1-TOM)的分层聚类来构建相关基因模块[12]。
1.3 重要模块的识别和功能分析 为确定各模块的生物学意义,以模块特征基因(MEs)为主成分筛选基因与临床性状的潜在相关性,总结各模块基因的表达模式。然后计算模块显著性(MS)与模块内的平均基因显著性(GS),衡量与样本性状(包括造模时长和疾病有无)相关的模块表达模式间的相关性[13]。一般来说,模块的MS 值越高则越重要[14]。使用R 软件的“clusterProfiler”包[15]对核心模块中的基因进行基因本体论(GO)和京都基因和基因组百科全书(KEGG)途径富集分析,以确定LN 的潜在发病机制和生物学途径。
1.4 DEGs 的鉴定与功能富集 利用R 语言的“LIMMA”软件包[16]设定|Log2FC|>1 且P<0.05 筛选LN 和正常肾脏组织样本间的DEGs。使用“cluster⁃Profiler”和“pathview”包[17]对差异基因进行GO 功能富集分析和KEGG信号通路分析。
1.5 LN高风险关键基因的筛选 高风险差异表达基因为WGCNA 筛选出的模块中的基因与DEGs 中的共同致病基因。
1.6 LN 关键基因的筛选 将筛选出的高风险DEGs 导入STRING 数据库构建PPI 网络,使用Cyto⁃scape3.7.2[18]将LN 高风险基因可视化,并分别使用插件“Cytohubba”[19]和“CytoNCA”[20]获取关键基因。
1.7 模型制备 雌性狼疮易感(SNF1)裸鼠20 只,体质量(18.0±0.7)g,购自北京维通利华实验动物有限公司并饲养于天津中医药大学实验动物中心[动物合格证号:SYXK(京)2016-0001]。选择体质量相近的裸鼠用随机数字表编号后,随机均分为对照组和模型组(n=10),适应性喂养1周后开始实验。采用单次腹腔注射0.5 ml降植烷的方法构建SLE模型[21]。对照组以0.5 ml/只单次腹腔注射PBS 溶液。每周定期观察裸鼠皮肤改变,3 个月后以裸鼠皮肤出现红斑样皮损、关节肿大及类风湿关节炎症状,血清抗dsDNA 抗体、抗C1q 抗体显著上升及补体C3、C4水平下降为造模成功[22]。
1.8 LN 肾脏组织总RNA 提取及RT-PCR 检测 采用RT-PCR 验证上述小鼠肾脏组织的4 个Hub 基因的表达变化。按照说明书用TRIzol试剂从肾脏组织中提取总RNA,采用PrimerScript ⅡFirst Strand cDNA 合成试剂盒反转录成cDNA。采取反应体系10 μl进行PCR 扩增,95 ℃5 min 预变性,95 ℃10 s,60 ℃30 s,45 个循环。熔解曲线65~95 ℃。利用美国国家生物信息中心(NCBI)Primer-BLAST 设计引物(序列见表1),由生工生物工程(上海)股份有限公司合成,以2-ΔΔCt法计算相对mRNA 表达量,并与对照组(β-actin mRNA表达量)进行比较。

表1 PCR引物序列Tab.1 PCR primer sequences
1.9 统计学分析 使用GraphPad Prism8.0 进行统计分析。对符合正态分布的两组数据进行t检验,P<0.05表示差异有统计学意义。
2 结果
2.1 共表达网络的构建 从GEO 数据库中下载GSE86425的原始数据集,使用R语言对原始数据进行背景校正及归一化等预处理,删除重复和未识别的基因后,共得到43 481个基因(图1A)。根据无尺度拟合指数R2首次为0.9 时确定了最佳软阈值为β=16(图1B),此时,网络的平均连接程度相对较高,能够包含足够的信息。将MEDissThres 设置为0.5以合并距离相近且相似的模块,设置最小模块的基因数目为30 个,剪切高度为0.25,生成86 个模块(图2)。其中red 模块中有3 902 个基因,darkred 模块有3 055 个基因,blue 模块有3 082 个基因,brown模块有2 884 个基因,灰色代表未被纳入任何模块的基因,故该模块被排除在进一步分析之外。

图1 样本聚类树及共表达网络的构建Fig.1 Construction of sample clustering trees and co-expres⁃sion networks

图2 筛选LN的共同表达模块Fig.2 Screening co-expression module of LN
2.2 模块间的相关性与重要模块的识别 为了寻找所有模块的共表达相似性,根据模块间的相关性计算特征基因,与其他模块相比,brown 模块与LN的有无呈正相关[0.44(P=0.000 01)](附图1,www.immune99.com),提示此模块中的基因可能在LN 的发生发展中起重要作用。图3 显示了棕色模块基因显著性(gene significance,GS)和模块成员(module membership,MM)相 关 系 数 为0.86(P<1e-200),因此,brown 模块被认为是与LN 疾病状态相关的重要模块。

图3 模块成员和基因重要性之间的相关性Fig.3 Correlation between module membership and gene importance
2.3 重要模块中基因的富集分析 使用cluster⁃Profiler 包对brown 模块中的基因进行GO 和KEGG富集分析,根据显著性程度,GO 功能富集分析与KEGG 通路分析均以P≤0.05 表示差异具有统计学意义。GO 富集分析共获得767 条GO 条目,结果显示,brown 模块中的蛋白主要通过T 细胞活化、细胞黏附的正向调节及细胞因子产生的正向调节等生物过程发挥作用。在细胞构成方面主要与细胞外基质和含胶原蛋白的细胞外基质有关;在分子功能方面主要涉及肌动蛋白结合、细胞黏附分子结合及细胞外基质结构成分等。分别从生物过程(biologi⁃cal processes,BP)、细胞成分(cellular component,CC)、分子功能(molecular function,MF)3 个层面选取P值排名前10 的功能信息,结果见图4A。KEGG通路注释分析结果显示,潜在靶点涉及319 条相关信息通路,经调研发现其中与LN 密切相关的信号通路有5 条,包括细胞因子-细胞因子受体相互作用、TNF 信号通路、Th17 细胞分化、Toll 样受体信号通路、NF-κB 信号通路、Th1 和Th2 细胞分化、NOD-类受体信号通路、T细胞受体信号通路、ECM 受体相互作用、IL-17 信号通路等。图4B 为P值较小的前20条相关通路信息。

图4 重要模块中基因的富集分析Fig.4 Enrichment analysis of genes in important modules
2.4 LN DEGs的筛选功能富集 数据集GSE78851包括54 例LN 肾脏标本和41 例正常小鼠肾脏标本,用LIMMA 软件包以P-value<0.05 和|logFC|≥1 为条件,共筛选出294 个DEGs,其中上调基因271 个,下调基因23 个(图5)。对294 个DEGs 进行GO 分析,主要涉及白细胞迁移、中性粒细胞趋化作用、细胞因子介导的信号传导途径、对防御反应的正向调节等生物过程(图6)。

图5 DEGs的筛选Fig.5 Screening of DEGs

图6 DEGs的GO富集分析Fig.6 GO enrichment analysis of DEGs
2.5 重要模块与DEGs 间关键基因的筛选 如图7A 所示,将DEGs 和brown 模块中的基因进行映射,获得了252 个LN 的相关基因,随后,将225 个交集基因导入STRING 数据库构建PPI 网络(图7B)。利用Cytoscape软件的CytoHubba插件MCC算法筛选出排名前20位的节点(图7C)。此外,采用CytoNCA插件中的Degree 算法得到排名前20 位的节点(图7D)。最终将两种算法获得的基因相互映射取交集,确定了4 个关键基因,即Hub 基因:Trim30a、Cxcl10、Irf7和Stat1(图7E)。
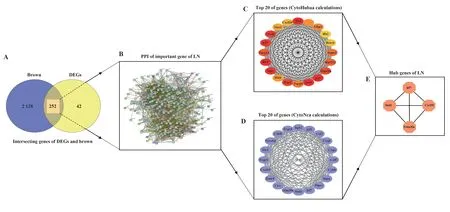
图7 Hub基因的筛选Fig.7 Screening of Hub genes
2.6 RT-PCR 验 证Hub 基 因 RT-PCR 分 别 验 证SNF1 裸鼠对照组和LN 组肾脏组织中的Hub 基因。结果表明,LN 小鼠肾脏Hub基因的mRNA 表达水平均显著高于正常对照组(P<0.01,图8)。

图8 Hub基因mRNA表达水平Fig.8 mRNA expression levels of Hub genes
3 讨论
LN 是SLE 最常见的表现之一,直接影响SLE 的预后,约40%的SLE患者伴有LN[2]。LN的病情活动及治疗效果可能会影响其预后[23],因此早期对LN患者病情进行准确地诊断及评估,有助于指导其临床治疗,提高治疗效果。本研究采用生物信息学方法,从LN 和正常肾脏组织中获取LN 的生物标志物及其病理过程。利用WGCNA 分析GSE86425 数据集,以探索与LN 临床特征相关的重要模块,结果获得86个模块,其中LN与brown模块的相关性最为显著。与其他生物信息学分析相比,WGCNA 可全面系统地计算出共表达模块与疾病临床特征间的关联,其筛选出的生物标志物具有高可靠性和准确性[24]。对brown 模块进行GO 功能注释分析发现,模块中蛋白主要富集于T 细胞活化、细胞因子产生的正向调节、细胞外基质结构成分等生物过程;关键模块蛋白的KEGG 通路分析也富集到了细胞因子-细胞因子受体相互作用、Th17 细胞分化、NF-κB 信号通路、Th1 和Th2 细胞分化及IL-17 信号通路,揭示机体内T细胞状态及炎症因子等免疫相关过程的改变,可能是LN 发生的重要环节。有研究表明,CD4+T 辅助性细胞(Th)在LN 等自身免疫性疾病的发生发展中发挥重要作用。Th1、Th17 等Th 细胞亚群和Treg 可通过诱导抑制其他免疫细胞增殖、分化和活化调节相关免疫反应[25]。LN 患者中Th1/Th2增高,高于健康对照组和无蛋白尿的SLE患者组,在有弥漫增殖性LN 的患者中这一倾向尤为明显[26]。KOMIYAMA 等[27]认为IL-17 表达量与LN 的严重程度密切相关,在LN 患者肾小球和肾间质浸润T细胞中可检测出大量IL-17[28]。IL-17 可通过刺激上皮细胞和成纤维细胞分泌趋化因子或炎症介质,导致炎症细胞浸润,从而参与LN 的发生发展[29]。基于此,T 细胞水平及炎症因子可能为LN 的治疗提供潜在靶点。
此外,对LN 和正常肾脏组织进行基因差异表达分析,筛选出294个DEGs。基于GO 富集分析,发现DEGs 参与白细胞迁移、中性粒细胞趋化作用、细胞因子介导的信号传导途径等。值得注意的是,白细胞表面标志物整合素Mac-1 和CD16b 被认为与LN的发病有关[30]。Mac-1包括一个独特的α亚单位(CD11b)与一个共同的β2 亚单位复合,在包括LN在内的炎症条件下与CD16b 一起从特定的白细胞亚群中释放出来[31]。KITAGAWA 等[32]发现体液中的CD11b 可能通过调节肾小球白细胞的积聚而参与LN的病理进展。
通过对WGCNA 重要模块的网络拓扑分析和DEGs 的综合分析,确定了Trim30a、Cxcl10、Irf7 和Stat1 4 个Hub 基因,并通过RT-PCR 验证。Trim30a隶属于TRIM 蛋白家族,是一种来自辅助性和诱导性T细胞的胞内蛋白,在免疫应答、代谢调控等方面均发挥重要作用,能够调节特殊细胞因子的核酸表达[33-34]。LN 患者体内免疫信号过度激活引发持续性炎症反应,损害宿主细胞。而Trim30 等天然免疫应答分子可通过相应的负反馈调节因子严格控制相关免疫通路的激活,进而保护机体不受损伤[35]。有研究人员发现Trim30a 可通过减少体内ROS 的产生控制NLRP3 受体的激活,使Caspase-1 和IL-1β 减少,避免LN 等疾病导致的过度的炎症反应给机体带来伤害[36]。CXCL10 为重要的免疫调节因子,可与受体结合后,激活CD4+的T 细胞、巨噬细胞及自然杀伤细胞,促使其向肾脏组织中积聚,诱发免疫相关炎症反应[37]。国内研究人员发现LN 患者血清中CXCL10 表达升高,促进病变进展[38]。干扰素调控基因7(Irf7)是通路的转录调节因子,是Ⅰ-IFN 通路的主要调控者。研究表明干扰素刺激基因及趋化因子的表达水平在LN 患者机体内显著升高,并且与肾炎活动性呈正相关[39]。肾脏的表达谱芯片亦提示LN 的肾脏组织中免疫细胞的活化等多条通路上调,包括Irf7 的表达水平较正常肾脏及单纯SLE 肾脏组织显著增高[40]。陈洁[41]发现Irf7 在LN患者肾中的蛋白水平和mRNA水平均有高表达,Irf7的肾小球表达积分与SLE 疾病的活动度、抗ds-DNA抗体、LN 的活动性评分呈现明显的正相关,而与血清C3、Ccr 呈负相关。以上均提示Irf7 分子可能是LN 发病的关键。Stat1 是信号转导子和转录激活因子(Stat)家族成员,常以非活性的形式存在于细胞质,活化后的Stat1 参与调控细胞的分化和凋亡等[42-43]。研究人员用LN 患者的血清分别刺激人肾小球系膜细胞和人肾小球内皮细胞,模拟LN 体内环境,发现IL-10 及TNF-α 等炎症因子表达增高,表明Stat1与LN 肾脏细胞释放促炎症因子的相关过程有关[44-45]。此外,临床研究也提示,LN患者体内T细胞中Stat1表达水平为单纯SLE患者的2.04倍,患者肾小管上皮细胞及肾小球中Stat1表达水平越高,预后则越差[46-47]。
综上所述,本文利用WGCNA、差异分析等多种生物信息学方法探讨了LN 可能的发病机制,富集到多个有意义的生物学过程和通路。其中,T 细胞活化、细胞因子产生的正向调节及白细胞迁移等过程的改变可能是LN 发生的重要环节。筛选出的4 个Hub 基因虽经过基础实验验证,但样本量较小,后续仍需大样本进行进一步研究,以筛选出灵敏性和特异性高的诊断标志物,为LN 的早期诊断和靶向药物研究提供重要参考。
猜你喜欢
中老年保健(2022年3期)2022-08-24
中老年保健(2022年3期)2022-08-24
老年医学研究(2021年5期)2022-01-19
昆明医科大学学报(2021年12期)2021-12-30
现代临床医学(2021年6期)2021-11-20
现代临床医学(2021年4期)2021-07-31
现代临床医学(2021年4期)2021-07-31
云南医药(2021年3期)2021-07-21
天津医科大学学报(2021年1期)2021-01-26
医学研究杂志(2015年12期)2015-06-10
