
3种淡水鱼病原菌多重PCR检测方法的建立与应用
2022-07-20 02:24:20胡宗云杨培民闫有利
水产科学 2022年4期
胡宗云,杨培民,闫有利
(辽宁省淡水水产科学研究院,辽宁省水生动物病害防治重点实验室,辽宁 辽阳 111000)

目前,水生动物致病菌的判定需要借助培养、分离和生化鉴定等技术[14-15],这种方法耗时费力,操作复杂且敏感性低;16S rDNA分子鉴定方法近年来应用较为广泛,这种方法需要测序,耗时长、费用高;此外,免疫学检测技术、单克隆抗体技术及单重PCR检测技术用于病原菌的检测也有报道,这些方法通常只能检测单种致病菌[16]。与单重PCR相比,多重PCR具有高效率、高通量及低成本的特性[17-18]。目前,多重PCR技术已被广泛用于基因突变[18]、病原微生物[19]、寄生虫[20]等多个领域的检测。就水生动物病原微生物检测而言,一些学者开发出了可同时检测多种致病菌[21-24]和病毒[25-26]的多重PCR检测方法;另有学者将多重PCR技术用于致病菌[27]和病毒[28]的分型研究。这些工作有效提升了检测的效率和准确性,在科研和生产中具有很大的应用潜力。笔者以近几年辽宁地区淡水鱼常见病原菌为检测对象,以嗜水气单胞菌的溶血素(hlyA)基因、迟钝爱德华氏菌的效应蛋白(eseD)基因、蜡样芽孢杆菌非溶血性肠毒素(nheA)基因为分子靶标,旨在建立可同时检测3种致病菌的单管多重PCR检测方法,以期为3种病原菌的快速诊断和分子流行病学调查提供技术支撑。
1 材料与方法
1.1 材料
1.1.1 试验菌株
嗜水气单胞菌AH10501、蜡样芽孢杆菌CMCC(B)63303和迟钝爱德华氏菌ATCC49231购自南京茂捷微生物科技有限公司;蜡样芽孢杆菌HHG[12]、爱德华氏菌(E.ictaluri)、维氏气单胞菌(A.veronii)、荧光假单胞菌(Pseudomonasfluorescens)、豚鼠气单胞菌(A.caviae)、希瓦氏菌(Shewanellasp.)、弗氏柠檬酸杆菌(Citrobacterfreundii)、类志贺邻单胞菌(Plesiomonasshigelloides)为本实验室保存菌株。
1.1.2 试剂与仪器
Taq DNA聚合酶、dNTP Mixture、10×PCR buffer、DL5000 marker购于宝生物工程(大连)有限公司,细菌基因组DNA提取试剂盒和动物组织基因组DNA提取试剂盒购于天根生化科技(北京)有限公司。主要仪器包括ABI 2720型PCR仪(赛默飞世尔科技,美国)、电泳仪(北京六一生物科技有限公司)、Alpha Imager HP凝胶成像系统(Protein Simple公司,美国)、Nano drop One C超微量紫外—可见光分光光度计(赛默飞世尔科技,美国)、高速冷冻离心机(贺默, 德国)。
1.2 方法
1.2.1 细菌培养与基因组DNA提取
挑取供试菌株接种于普通营养琼脂平板上,37 ℃恒温培养24 h后,用接种环挑取单克隆菌落置于灭菌的1.5 mL EP管中,按照试剂盒操作说明提取基因组DNA。提取的DNA经Nano Drop One C测定浓度后,置于-20 ℃冰箱中储存备用。
1.2.2 引物设计与合成
根据GenBank中登录的3个细菌对应的毒力基因序列,通过Primer primier 6.0软件,并参考已发表的文献,设计多对引物;引物由生工生物工程(上海)股份有限公司合成;经NCBI数据库BLAST比对和预试验验证后获得了3对特异性和兼容性较强的引物对(表1)。
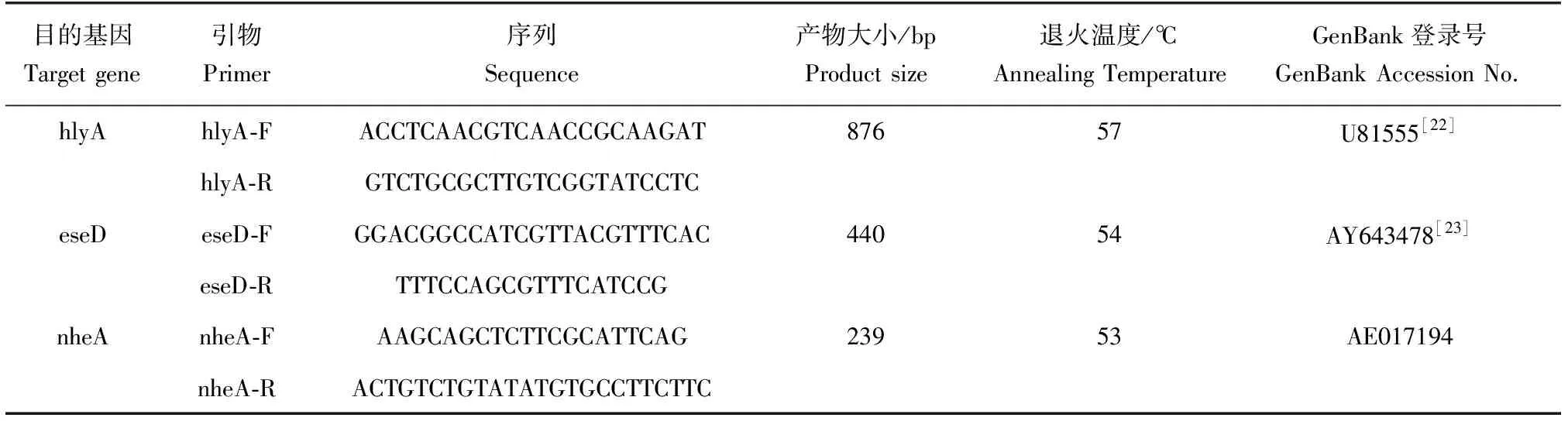
表1 多重PCR扩增用引物信息
1.2.3 多重PCR特异性检验
取各待测细菌基因组DNA 50 ng作为模板,加入25 μL反应体系中进行特异性检验。反应体系包括:hlyA、eseD和nheA引物3对(每条引物终浓度0.4 μmol/L),10×PCR缓冲液(含Mg2+)2.5 μL,dNTP混合物(各2.5 mmol/L)2 μL,rTaq酶(5 U/μL)0.3 μL,最后用灭菌双蒸水补足25 μL。PCR反应程序为:94 ℃预变性5 min;94 ℃变性1 min,55 ℃退火40 s,72 ℃延伸1 min,30个循环;最后72 ℃延伸7 min。PCR产物在1%琼脂糖凝胶电泳检测(90 V电压下电泳30 min),选择嗜水气单胞菌AH10501、蜡样芽孢杆菌CMCC(B)63303和迟钝爱德华氏菌ATCC49231的PCR产物送至生工生物工程(上海)股份有限公司进行测序,将所得序列与GenBank中的菌株进行比对。
1.2.4 多重PCR反应体系的建立和优化
1.2.4.1 模板质量浓度组合的优化
在25 μL PCR反应体系中,将3株标准菌株的基因组DNA按不同质量浓度进行组合,组合中嗜水气单胞菌AH10501、蜡样芽孢杆菌CMCC(B)63303和迟钝爱德华氏菌ATCC 49231的终质量浓度分别为1.0、2.5、2.5 ng/μL,2.5、1.0、1.0 ng/μL,1.0、2.5、1.0 ng/μL,2.5、2.5、2.5 ng/μL和1.0、1.0、1.0 ng/μL。PCR反应体系其他组分为:3个基因的上、下游引物各1 μL(每条引物终浓度为0.4 μmol/L),10×PCR缓冲液(含Mg2+)2.5 μL,dNTP混合物(各 2.5 mmol/L)2 μL,rTaq酶(5 U/μL)0.3 μL,最后用双蒸水补足25 μL。反应程序同1.2.3,PCR产物经1%琼脂糖凝胶电泳检测,根据条带亮暗及清晰程度确定最佳的模板质量浓度组合。
1.2.4.2 引物浓度的优化
在25 μL PCR反应体系中,通过添加不同体积的引物使每条引物的终浓度分别为0.2、0.4、0.6、0.8 μmol/L,反应体系与反应程序同1.2.3。PCR产物经1.0%琼脂糖凝胶电泳检测,分析引物浓度对多重PCR的影响。
1.2.4.3 dNTP浓度的优化
在25 μL反应体系中,dNTP添加量分别为0.5、1.0、1.5、2.0、2.5、3.0 μL,对应的dNTP总浓度分别为0.05、0.10、0.15、0.20、0.25、0.30 μmol/L,其余组成及反应程序与1.2.3相同。通过筛选试验确定dNTP最适浓度。
1.2.4.4 酶活性对多重PCR的影响
在25 μL反应体系中,调整酶的添加量使其总活性分别为0.02、0.03、0.04、0.05、0.06、0.07 U/μL, 其余组成及反应程序与1.2.3相同。PCR产物经1%琼脂糖电泳检测,获得酶的最佳用量。
1.2.4.5 退火温度的优化
25 μL反应体系中各组分与1.2.3相同,PCR反应程序为:94 ℃预变性5 min;94 ℃变性1 min,分别在51、53、55、57、59 ℃和61 ℃退火40 s;72 ℃延伸1 min,30个循环;最后72 ℃延伸7 min。PCR产物经1%琼脂糖凝胶电泳检测,筛选出最佳的退火温度。
1.2.5 多重PCR敏感性检测
将3株标准菌株的基因组DNA从最初浓度开始按10倍倍比进行稀释,将不同稀释度的细菌基因组DNA分别取1 μL加入反应体系,作为模板进行多重PCR扩增,以检测多重PCR反应的灵敏度。
1.2.6 多重PCR临床应用
应用本次建立的多重PCR检测方法,对本实验室近年来从常见淡水鱼病鱼鱼体中分离、保存的218株菌株进行检测。同时对218株菌株进行细菌基因组DNA提取、PCR扩增16S rDNA基因并送到生工生物工程(上海)股份有限公司进行测序,所得16S rDNA序列在NCBI数据库中进行BLAST比对、同源性分析,进行菌株的分子鉴定。比较多重PCR检测方法和16S rDNA测序鉴定这两种方法所得结果是否一致。
2 结 果
2.1 多重PCR特异性分析
不同模板在多重PCR检测体系中的扩增情况见图1,1~4泳道分别为3个标准菌株和本实验室分离的蜡样芽孢杆菌HHG株,分别扩增出900、500 bp和250 bp大小的片段,PCR产物片段长度与预测基本一致。而其他参考菌株或宿主基因组DNA做模板时无PCR扩增产物,表明本试验所建立的多重PCR体系与其他菌株及宿主基因组无扩增反应,该体系具有一定的特异性。
扩增产物经测序并与NCBI数据库中对应的序列进行比对发现,hlyA、eseD和nheA 3个基因与GenBank中的嗜水气单胞菌hlyA基因(U81555)、迟钝爱德华氏eseD基因(AY643478)和蜡样芽孢杆菌nheA基因(AE017194)高度同源,同源率超过99%。这表明,本体系所扩增的产物均为目的产物。
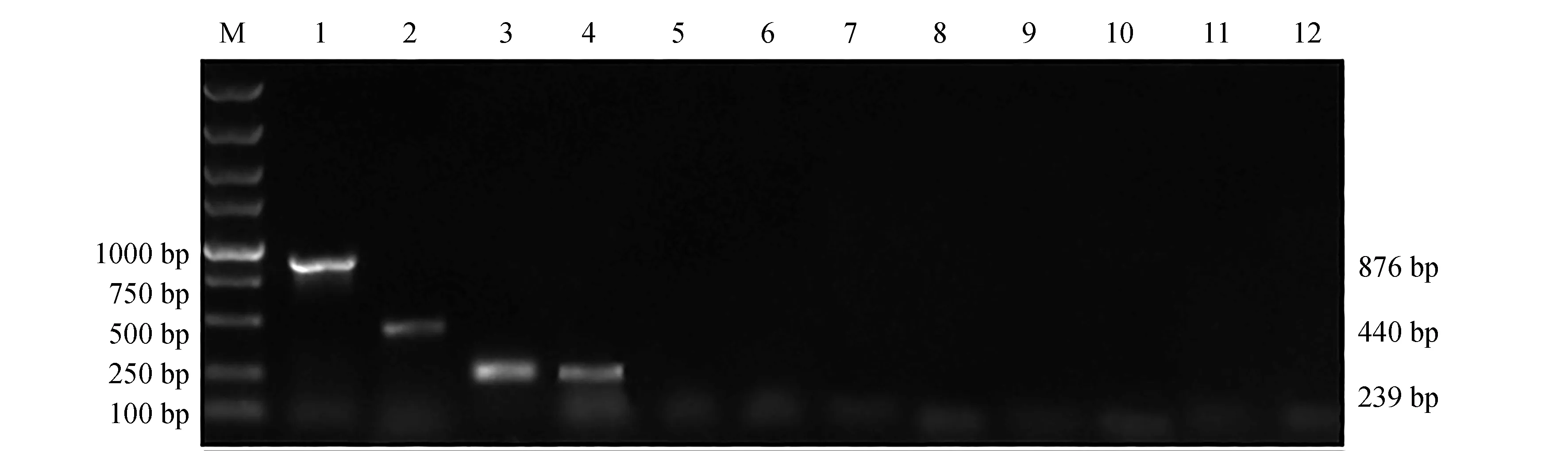
图1 不同模板在多重PCR反应检测体系扩增状况Fig.1 Agarose gel electrophoresis of multiplex PCR amplified against various DNA templatesM.DL 5000 DNA marker; 1.嗜水气单胞菌AH10501; 2.迟钝爱德华氏菌ATCC49231; 3.蜡样芽孢杆菌CMCC(B)63303; 4.蜡样芽孢杆菌分离株HHG; 5.维氏气单胞菌; 6.豚鼠气单胞菌; 7.希瓦氏菌; 8.类志贺邻单胞菌; 9.弗氏柠檬酸杆菌; 10.荧光假单胞菌; 11.鲤基因组DNA; 12.空白对照.M.DL 5000 marker; 1.A. hydrophila AH10501; 2.E. tarda ATCC49231; 3.B. cereus CMCC(B)63303; 4.B. cereus HHG; 5.A. veronii; 6.A. caviae; 7.Shewanella sp.; 8.P. shigelloides; 9.C. freundii; 10.P. fluorescens; 11.genome DNA of common carp Cyprinus carpio; 12.the blank control.
2.2 多重PCR体系的建立和优化
2.2.1 模板质量浓度组合优化
在多重PCR体系中3对引物终浓度均为0.4 μmol/L基础上,3种菌株基因组DNA不同质量浓度组合的PCR产物电泳结果见图2。3个菌株基因组DNA终质量浓度为1.0 ng/μL时,在各个组合中扩增效率均较低;而终质量浓度达到2.5 ng/μL时,扩增效率明显增强。因此,3个菌株基因组DNA最佳模板质量浓度组合为2.5、2.5、2.5 ng/μL。
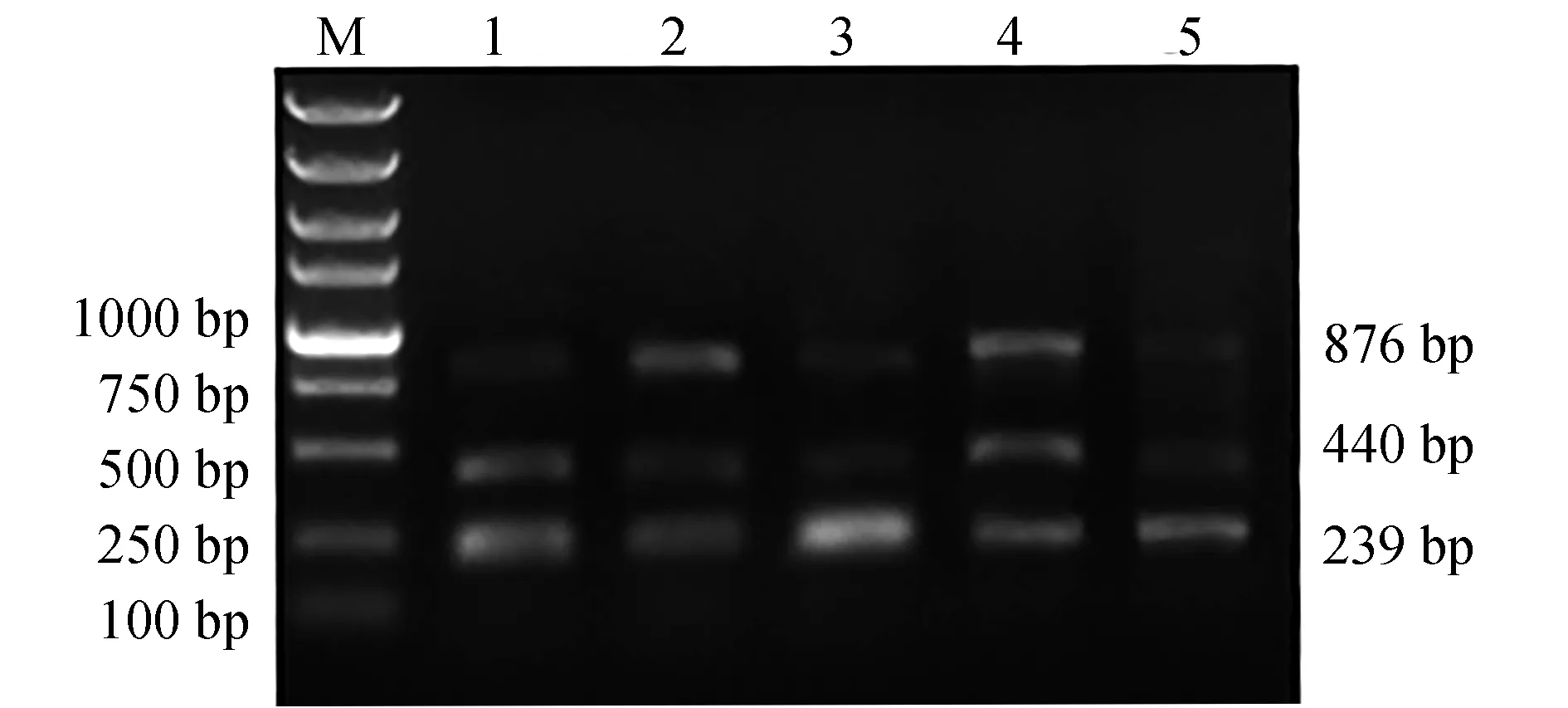
图2 不同模板质量浓度组合对多重PCR结果的影响Fig.2 Agarose gel electrophoresis of multiplex PCR amplified against different DNA concentrations among three pathogensM.DL 5000 DNA marker;1~5.嗜水气单胞菌、蜡样芽孢杆菌和迟钝爱德华氏菌基因组DNA模板质量浓度分别为1.0、2.5、2.5 ng/μL,2.5、1.0、1.0 ng/μL,1.0、2.5、1.0 ng/μL,2.5、2.5、2.5 ng/μL,1.0、1.0、1.0 ng/μL.M.DL5000 marker; 1—5.the DNA concentrations of three pathogens (A. hydrophila, B. cereus and E. tarda) in multiplex PCR are 1.0, 2.5, 2.5 ng/μL, 2.5, 1.0, 1.0 ng/μL, 1.0, 2.5, 1.0 ng/μL, 2.5, 2.5, 2.5 ng/μL, and 1.0, 1.0, 1.0 ng/μL, respectively.
2.2.2 引物浓度优化
调整引物浓度进行多重PCR反应,结果见图3。当引物浓度为0.2~0.8 μmol/L时,hlyA和nheA基因扩增效率变化不大;但eseD基因在引物浓度为0.4 μmol/L时扩增效率最高。因此,多重PCR体系最适引物浓度为0.4 μmol/L。
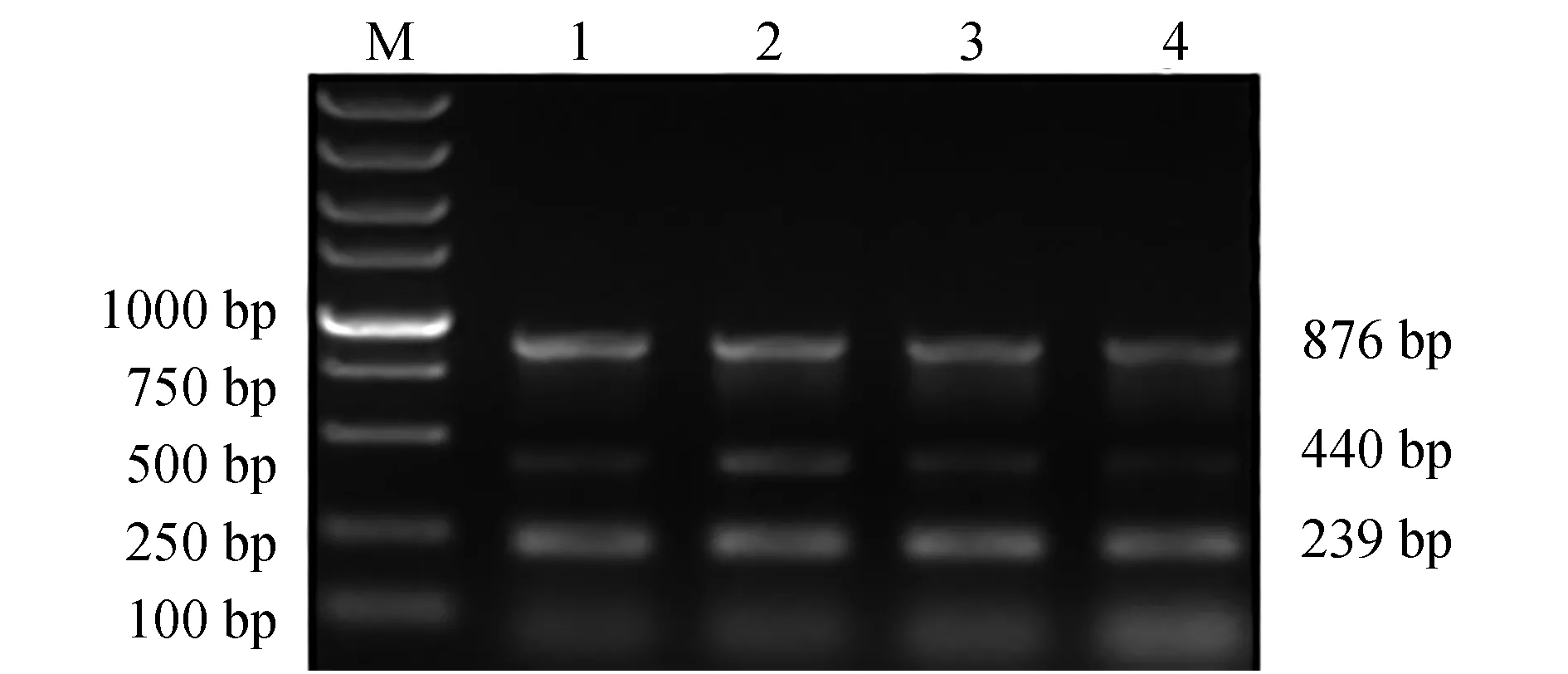
图3 多重PCR最适引物浓度的选择Fig.3 Optimization of primer concentration for multiplex PCR detection systemM.DL 5000 DNA marker; 1~4.引物终浓度分别为0.2、0.4、0.6、0.8 μmol/L.M.DL 5000 DNA marker; 1—4.the primer concentration of multiplex PCR is 0.2, 0.4, 0.6 and 0.8 μmol/L, respectively.
2.2.3 dNTP浓度优化
dNTP浓度变化对多重PCR体系的影响见图4。25 μL反应体系中,当dNTP终浓度为0.20 μmol/L时,3个基因PCR产物条带最亮,扩增效率最高。因此,dNTP最适终浓度为0.20 μmol/L,所对应的dNTP添加量为2.0 μL。

图4 多重PCR 最适dNTP终浓度的选择Fig.4 Optimization of dNTP concentration for multiplex PCR detection systemM.DL 5000 DNA marker; 1~6.dNTP终浓度分别为0.05、0.10、0.15、0.20、0.25、0.30 μmol/L.M.DL 5000 DNA marker; 1—6.the concentration of dNTP is 0.05, 0.10, 0.15, 0.20, 0.25 and 0.30 μmol/L, respectively.
2.2.4 多重PCR体系中酶浓度的优化
在25 μL多重PCR体系中,酶活性由0.02 U/μL增至0.05 U/μL时,3个基因扩增效率无明显差异;当酶活性为0.06、0.07 U/μL时,3个基因扩增效率较高,其中酶活性为0.06 U/μL时,条带清晰,可满足多重PCR检测体系的需要(图5)。

图5 多重PCR最适酶活性的选择Fig.5 Optimization of DNA polymerase activity for multiplex PCR detection systemM.DL 5000 DNA marker; 1~6.酶活性分别为0.02、0.03、0.04、0.05、0.06、0.07 U/μL.M.DL 5000 DNA marker; 1—6.the activity of DNA polymerase is 0.02, 0.03, 0.04, 0.05, 0.06 and 0.07 U/μL, respectively.
2.2.5 多重PCR体系中退火温度的优化
当退火温度在51~61 ℃变化时,3个基因均可获得有效的扩增(图6)。当退火温度为51 ℃和57 ℃时,3个基因扩增效率相对较低;当退火温度为59 ℃和61 ℃时,hlyA和nheA基因扩增效率略高于eseD基因;当退火温度为53 ℃和55 ℃时,3个基因扩增效率较高,条带清晰明亮,为避免温度较低引起非特异扩增,选择55 ℃作为多重PCR反应体系的退火温度。
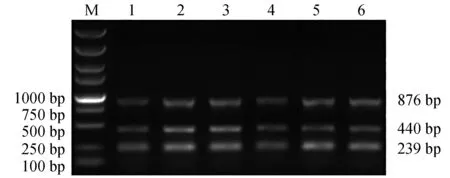
图6 多重PCR 最适退火温度的选择Fig.6 Optimization of annealing temperature for multiplex PCR detection systemM.DL 5000 DNA marker; 1~6.退火温度分别为51、53、55、57、59、61 ℃.M.DL 5000 DNA marker; 1—6.the annealing temperature is 51, 53, 55, 57, 59 and 61 ℃, respectively.
多重PCR反应条件优化结果表明,最佳反应体系为:hlyA、eseD和nheA基因上、下游引物(10 μmol/L)各1.0 μL,嗜水气单胞菌AH10501、蜡样芽孢杆菌CMCC(B)63303和迟钝爱德华氏菌ATCC49231 DNA添加量均为50 ng,10×PCR缓冲液(含Mg2+)2.5 μL,dNTP混合物(各2.5 mmol/L)2 μL,rTaq酶(5 U/μL)0.3 μL,最后用灭菌双蒸水补足25 μL。最佳反应条件为:94 ℃预变性5 min;94 ℃变性1 min,55 ℃退火40 s,72 ℃延伸1 min,30个循环;最后72 ℃延伸7 min。
2.3 多重PCR敏感性检测
本试验中试剂盒提取的嗜水气单胞菌AH10501、迟钝爱德华氏菌ATCC49231和蜡样芽孢杆菌CMCC(B)63303基因组DNA质量浓度分别为48、30 ng/μL和26 ng/μL。当稀释倍数为10时,模板添加量为4.8、3.0 ng和2.6 ng,hlyA、eseD和nheA基因均有可识别的较弱扩增。因此,在本试验的设计中,多重PCR反应体系对嗜水气单胞菌、迟钝爱德华氏菌和蜡样芽孢杆菌最低检出剂量分别为4.8、3.0 ng和2.6 ng(图7)。
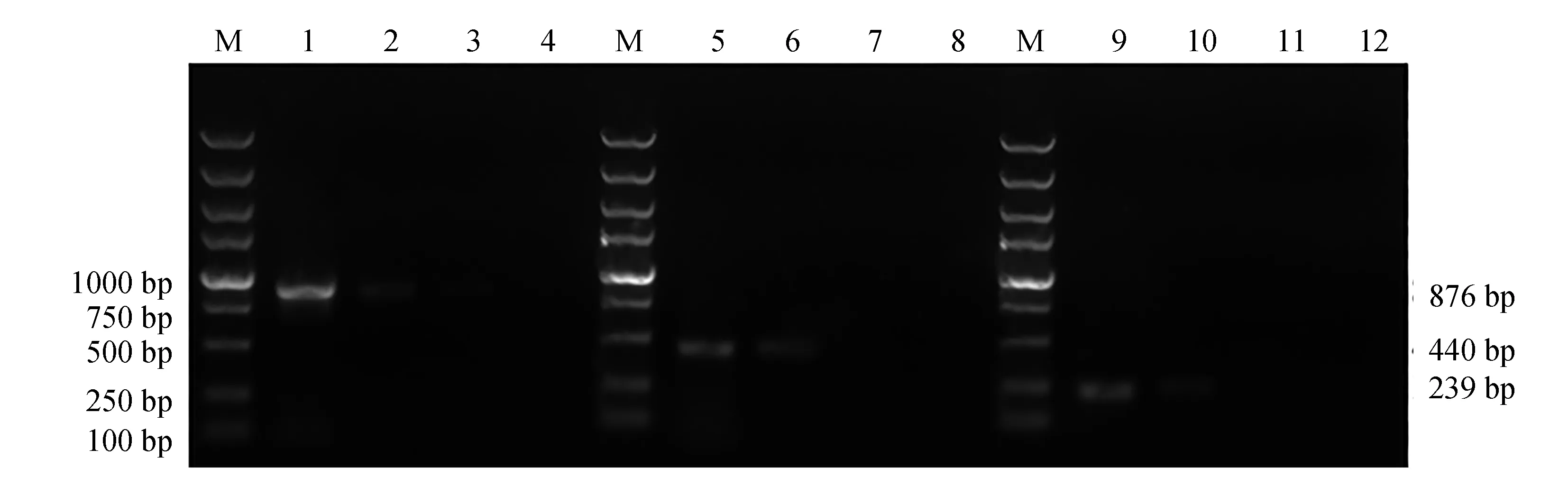
图7 模板稀释法测定多重PCR敏感性试验Fig.7 Sensitivity assay of multiplex PCR by DNA templates dilutionM.DL 5000 DNA marker; 1~4.嗜水气单胞菌AH10501基因组DNA 100~103稀释后作为模板扩增状况; 5~8.迟钝爱德华氏菌ATCC49231基因组DNA 100~103稀释后作为模板扩增状况; 9~12.蜡样芽孢杆菌CMCC(B)63303基因组DNA 100~103稀释后作为模板扩增状况.M.DL 5000 DNA marker; 1—4.agarose gel electrophoresis of multiplex PCR amplified against various DNA templates ranging from 100—103 in A. hydrophila AH10501; 5—8.agarose gel electrophoresis of multiplex PCR amplified against various DNA templates ranging from 100—103 in E. tarda ATCC49231; 9—12.agarose gel electrophoresis of multiplex PCR amplified against various DNA templates ranging from 100—103 in B. cereus CMCC(B)63303.
2.4 多重PCR临床应用
应用本次建立的多重PCR方法,对本实验室近年来从各种病鱼鱼体中分离、保存的218株菌株进行检测。经多重PCR方法检测发现,阳性样本187个,其中嗜水气单胞菌108株、蜡样芽孢杆菌54株、迟钝爱德华氏菌25株;剩余31株DNA未扩增目的条带(图8)。同时对218株菌株进行16S rDNA基因测序验证,所得16S rDNA序列在美国国家生物技术信息中心中进行BLAST比对及同源性分析,结果显示,108株与嗜水气单胞菌、25株与迟钝爱德华氏菌、54株与蜡样芽孢杆菌的同源性达到99%以上,其他31株,主要为维氏气单胞菌和柱状嗜纤维菌(Cytophagacolumnaris)、弗氏柠檬酸杆菌等。多重PCR检测分离菌株与16S rDNA鉴定结果进行比较,结果显示,2种方法均鉴定出嗜水气单胞菌108株、迟钝爱德华氏菌25株、蜡样芽孢杆菌54株,符合率达100%(表2)。
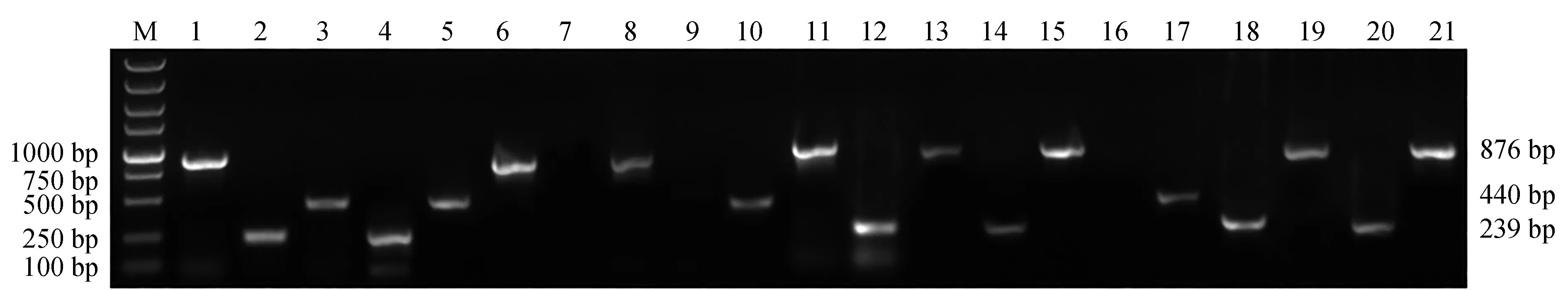
图8 多重PCR体系检测分离菌株的电泳结果Fig.8 Agarose electrophoresis of multiplex PCR amplified by the template of the isolated strainsM.DL 5000 DNA marker; 1~21.分离菌株基因组DNA在多重PCR体系中扩增结果.M.DL 5000 DNA marker; 1—21.agarose electrophoresis of multiplex PCR amplified by the sample template in genomes of the isolates.
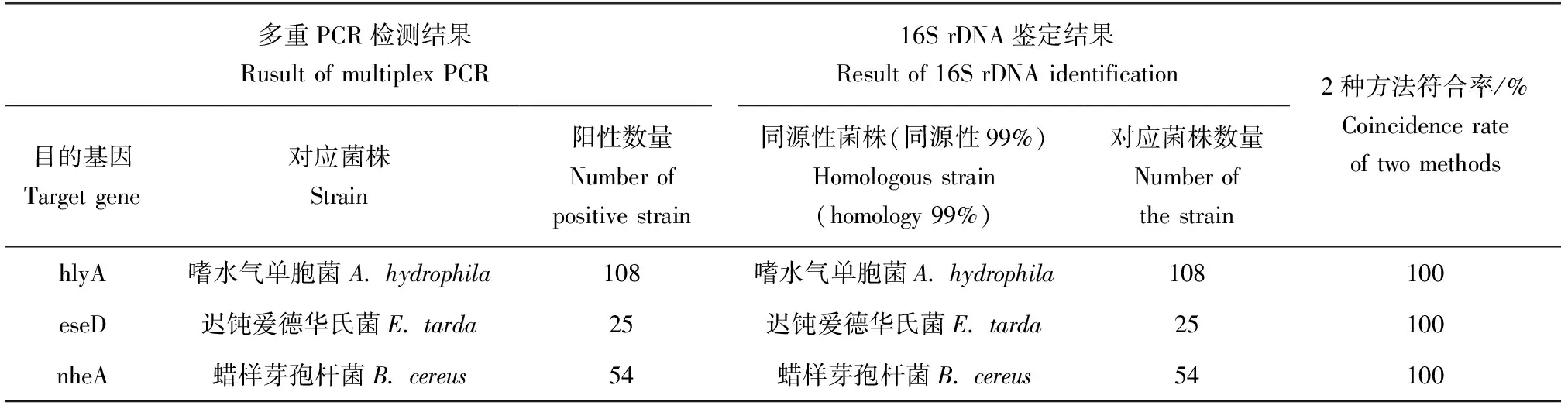
表2 多重PCR检测分离菌株与16S rDNA鉴定结果比较
3 讨 论
3.1 影响多重PCR特异性扩增的因素
聚合酶链式反应(PCR)技术是一种体外扩增特异DNA片段的分子生物学技术,可将微量的基因片段短时间内进行指数扩增,并具有较高的灵敏度和特异性,可对目的基因进行快速检测,目前广泛地应用于水产动物疾病检测中[29-30]。多重PCR又称多重引物PCR,是在同一PCR反应管中同时加上多种病原微生物的特异性引物进行PCR扩增。多重PCR检测方法的关键是引物设计,引物要特异性强,引物内部避免形成发卡结构,在扩增过程中引物与引物之间避免形成二聚体,扩增的PCR产物大小不同且能在电泳时区分开。笔者以嗜水气单胞菌的溶血素(hlyA)基因、迟钝爱德华氏菌的效应蛋白(eseD)基因、蜡样芽孢杆菌非溶血性肠毒素(nheA)基因为分子靶标,合成了3对特异性引物,以3种菌的混合DNA为模板,运用多重PCR技术成功扩增出了长度分别为876、440 bp和239 bp的目的片段,通过特异性检验分析了该反应体系在不同菌株中的扩增状况,确定了该多重PCR检测体系的特异性。通过对模板DNA浓度、引物浓度、dNTP浓度、Taq酶活性和退火温度等进行多重PCR条件的优化,建立了最适的PCR反应体系,并用建立的多重PCR体系对本实验室分离菌株进行鉴定,其结果与16S rDNA鉴定结果的符合率为100%。该PCR检测体系的建立有效缩短了3种鱼源致病菌的检测时间,简化了操作过程,降低了检测成本,可以满足临床检测的需要。
3.2 分子靶标的选择
针对辽宁地区常见鱼源嗜水气单胞菌、迟钝爱德华氏菌、蜡样芽孢杆菌的相关毒力因子,以嗜水气单胞菌的hlyA基因、迟钝爱德华氏菌的eseD基因、蜡样芽孢杆菌的nheA基因为分子靶标。hlyA基因是嗜水气单胞菌的重要毒力因子,大多数气单胞菌属于条件性致病菌,具有致病菌株和非致病菌株之分,以往的研究已证实,hlyA基因可作为分子靶标进行致病性气单胞菌的鉴定[31-32],所检测的非致病性嗜水气单胞菌分离株均未扩增出hlyA和气溶素(aerA)基因,而致病性嗜水气单胞菌分离株中至少含有hlyA基因。eseD基因是致病性迟钝爱德华氏菌胞外产物的重要成分,在致病过程中起着重要作用,并在大菱鲆迟钝爱德华氏菌毒力因子检测中有所报道[33-35]。nheA基因在蜡样芽胞杆菌和苏云金芽孢杆菌(B.thuringiensis)中广泛存在[36],常作为分子靶标用于蜡样芽孢杆菌鉴定和分子分型[37-39]。目前,有关水生动物源致病性蜡样芽孢杆菌多重PCR检测的报道相对较少,一些学者多采用单重PCR[40-41]或测序[13]的方式研究蜡样芽孢杆菌毒力因子的分布情况。本试验中,在多株蜡样芽孢杆菌中检测到了nheA基因,该基因也存在于引发中华鳖“摇头病”[40]和半滑舌鳎(Cynoglossussemilaevis)出血症[41]的蜡样芽孢杆菌中,而通过测序可知,大菱鲆源蜡样芽孢杆菌基因组序列[13]中不存在nheA基因。此差异是由菌株生存环境或来源不同引起的还是由研究方法不同造成的有待进一步研究。
3.3 多重PCR检测极限的比较
本试验中采用模板稀释法来确定多重PCR体系的最低检测剂量,这种方法在鳗弧菌 (Vibrioanguillarum)[42]、致病性嗜水气单胞菌[31]的多重PCR检测中也有应用。一些学者将菌液梯度稀释后再提取DNA进行检测[17,43],菌液稀释后提取DNA的量受菌液混匀程度、试剂盒的效率、个人操作等多种因素影响,由此获得的最低检出量存在较大误差的可能性将会增加,不利于不同研究间检测极限的比较。
病原菌的毒力大小与其基因型有关,饶静静等[31]研究发现,所检测的非致病性嗜水气单胞菌分离株均未扩增出hlyA和aerA基因,而致病性嗜水气单胞菌分离株中至少含有hlyA基因,其建立的多重PCR体系对hlyA基因的最低可检测模板量为10 ng。笔者建立的多重PCR体系对hlyA检测极限为4.8 ng,高于李晨等[23]报道的最低检测水平(256 pg)。这表明本试验所建立的多重PCR体系对致病性嗜水气单胞菌的检测是有效的和灵敏度适中的。
4 结 论
针对辽宁地区常见淡水鱼病原菌嗜水气单胞菌、迟钝爱德华氏菌、蜡样芽孢杆菌的相关毒力因子,选择特异性较强的嗜水气单胞菌的溶血素(hlyA)基因、蜡样芽孢杆菌非溶血性肠毒素(nheA)基因、迟钝爱德华氏菌的效应蛋白(eseD)基因为分子靶标,分别设计1对特异性引物,并对反应体系进行条件优化,建立可同步检测3种菌的多重PCR检测体系。结果显示,3个毒力基因引物浓度均为0.4 μmol/L,模板质量浓度为2.5 ng/μL, dNTP和酶添加量分别为2.5 μL和0.3 μL,退火温度为55 ℃时,各目的片段均可较好地扩增。敏感性试验结果表明,建立的多重PCR对嗜水气单胞菌、迟钝爱德华氏菌和蜡样芽孢杆菌的最低检测剂量分别为4.8、3.0、2.6 ng。对临床分离的菌株样本进行检测,与16S rDNA分子鉴定结果的符合率为100%。
笔者建立的多重PCR体系检测方法特异、灵敏、快速,可同步完成淡水鱼源的嗜水气单胞菌、迟钝爱德华氏菌和蜡样芽孢杆菌的快速检测、鉴定,增强了本地区鱼病流行病学调查的准确性和可靠性。
猜你喜欢
中国人兽共患病学报(2024年2期)2024-03-15 02:41:52
生命与灾害(2019年7期)2019-08-07 03:36:34
中成药(2017年9期)2017-12-19 13:34:21
中华老年口腔医学杂志(2016年2期)2017-01-15 14:24:48
中国酿造(2016年12期)2016-03-01 03:08:09
动物营养学报(2015年10期)2015-12-01 02:26:34
现代检验医学杂志(2015年1期)2015-02-06 01:59:05
食品工业科技(2014年23期)2014-03-11 18:19:08
华东理工大学学报(自然科学版)(2014年5期)2014-02-27 13:49:27
食品科学(2013年15期)2013-03-11 18:25:40
