
通过分子动力学方法研究沥青质聚集体应变下势能变化
2022-07-19 03:49叶正扬樊小哲代振宇
石油学报(石油加工) 2022年4期
叶正扬, 樊小哲, 龙 军, 代振宇
(中国石化 石油化工科学研究院,北京 100083)
石油沥青是一种重要的道路交通材料,其性能直接决定了沥青路面的工程质量和结构形式。和水泥混凝土刚性路面不同,沥青路面属于柔性路面,相比具有行车舒适平稳、燃油消耗和机械磨损低的优点[1]。因此,改进沥青材料性能,研究沥青结构与性能之间的关系兼有经济价值和战略意义。目前,改进沥青材料性能的主要方法是添加其他物质。最常见的添加物是SBS胶粉等高分子物质[2],或添加石油树脂等其他石油组分进行调和实现改性[3],另外也有通过添加碳纳米管等新型碳基材料进行沥青改性的研究[4]。尽管这些改性方法都不同程度地实现了沥青改性需求,但没有研究沥青材料自身组成结构与性能关系。
沥青各相展现的形变状态、刚度和设计允许形变范围内的强度均与分子形变和分子间相互作用有关:对沥青材料施加外力时,沥青中分子间距和分子构型发生改变使材料整体应变,从而通过应力对抗外部作用。研究不同结构的分子在不同环境应变下的势能变化情况,可以体现材料整体形变下分子结构和相互作用的变化,由此揭示不同结构分子对材料内应力的贡献,从而为提升沥青材料使用性能提供理论依据和方向。
沥青的分子组成包含数万种分子,复杂的分子组成导致其内部结构的复杂性。通常对沥青化学成分的分析仅停留在拟组分分析,例如Corbett分析法[5],这些方法难以关联沥青组成参数和材料性能之间的关系。通过原子力显微镜观察发现,沥青材料并非均相体系,而是在微米尺度由力学性质完全不同的多个相组成,这些由不同分子组成的相之间相互交错渗透[6]。以沥青质为例,在若干纳米的尺度上具有自发相互聚集的特点,无论在减压渣油[7]或小分子溶剂[8]中均可形成具有分形结构的纳米聚集体,这些聚集体相互连接呈100 nm左右的小微粒,最终形成介观尺度的分相状态。
沥青在外力作用下的形变情况也相对复杂,在介观尺度展现出黏弹性。原子力相探测显微镜实验显示,沥青中各介观相在输入的正弦外力信号作用下会输出具有不同相位差的应变信号,这说明各相形变中弹性形变和塑性形变贡献有所区别。常温下沥青主要分为3种介观相:高低错落相(Catana-phase,一些文献中称为蜂型结构,本文根据其载荷形变响应特征称为高贮能模量相)、周相(Peri-phase)和邻相(Para-phase)。高贮能模量相结构相对有序,主要由沥青质组成,载荷下主要发生弹性形变;周相主要由软沥青中的芳香化合物构成,围绕高贮能模量相分布,强度相对较大;邻相主要由饱和分构成,柔软且塑性形变特征最明显[9]。各介观相受力形变的响应特征不同,增加了宏观尺度研究沥青组成与材料性能关系的难度。
基于沥青质自发相互聚集的特性,沥青质富集在沥青材料以弹性形变为主的高贮能模量相中,在材料整体中起弹性骨架作用[10]。通过分子动力学模拟的方法,构建无定形动力学盒子并将不同的沥青质模型分子分别放入盒中,在模拟真实环境的温度、压强下构建沥青质聚集体模型,模拟实际条件下沥青质的存在环境。对这些盒子施加应变后统计分子构型和相互作用的变化,实现从分子尺度分析沥青在形变过程中各种因素的贡献,为开发更高性能的沥青产品提供理论基础,也为后续的研究提供可行的方案。该方法也可以扩展到沥青其他介观相分子的形变分析中。
沥青质分子间的范德华作用和静电相互作用由分子动力学结果直接得出。但由于模拟体系中的沥青质分子数量较多且分子结构复杂,如果将所有分子的每一个键长、键角、扭矩参数全部统计并直接用于描述每个分子的构型,巨大的数据量显然难以统计,也无法直观统计应变下众多分子的形变情况。因此笔者采用统计每个盒子中沥青质分子的形变相关势能,通过相关各项势能值的相对变化对分子的伸缩、键角变化和扭曲等形变类别进行统计,从而描述沥青质分子在实际聚集状态下随形变发生时的构型变化。
1 研究对象与分子动力学模型构建
1.1 沥青质模型分子
沥青质是沥青中不溶于正构烷烃(正戊烷或正庚烷)而溶于甲苯的拟组分。沥青质作为一类相对分子质量在500~1500的稠环芳香化合物,分子种类繁多,因此无法对每种分子进行分离表征,但结构具有一定的共性,可以用Mullins的“岛式结构”模型加以概括[11]。沥青质分子结构可以概括为几大特征要素,即相对分子质量、稠环连接方式、杂原子种类与含量、侧链数量(即大小)等,这些结构因素可以概括不同结构类型沥青质分子的差异。
沥青质稠环核心的芳环缩合方式分为渺位缩合(cata-Condensed)和迫位缩合(peri-Condensed)2种,缩合方式除了决定稠环核心的构型外,也能影响分子的结构稳定性和稠环的共轭电子分布[12-13]。因此笔者以这一结构特征作为选择沥青质模型分子的标准。
以3种沥青质分子作为模型化合物,分别记作As1、As2和As3,分子结构如图1所示。3种沥青质均来自石油沥青,分子精细结构由Schuler等[14]通过原子力显微镜实验扫描获得。As1、As2分子符合大气压光电离傅里叶变换离子回旋共振质谱表征出的高含量沥青质的结构特征,As3则相对含量较少,沥青荧光发射光谱也同样支持这一结论[15-16]。3种分子均为C50左右。As1的芳环全部为渺位缩合,带有2个较短的侧链;As2带有1个由6个芳环迫位缩合而成的单元,其他芳环均为渺位缩合,并有若干个甲基取代基;As3则为高度迫位缩合的沥青质,碳/氢原子比相比前2种分子明显更高,且不含取代侧链。
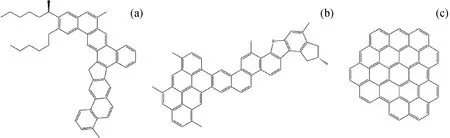
图1 3种沥青质分子结构模型Fig.1 Model molecules of three kinds of asphaltenes(a) As1—Totally cata-condensed asphaltene; (b) As2—Partly cata-condensed asphaltene; (c) As3—Totally peri-condensed asphaltene
1.2 沥青质聚集体及形变特点
沥青分子的不均匀分布有别于传统的胶束模型,沥青中各相遵循溶胶-凝胶(Sol-Gel)结构。对于沥青质而言,4~8个沥青质分子先形成盘状的纳米聚集体[8,17],这些聚集体又进一步聚集形成100 nm以下、维数1.7左右的分形簇。介观尺度上,部分分型簇“悬浮”在周相内部,形成类似胶束的结构;另一些则相互聚集形成连续结构并和其他相相互渗透,形成相边界复杂的分形结构,HausDorff维数大约在1.7~1.9[10]。分子动力学的模拟尺度可以满足模拟分形簇的要求,但远小于分相出现的介观尺度。
沥青质所在的相以弹性形变为主,但依然具有黏弹性形变特征,不能简单将该相形变特征等同于晶体等周期型结构,对比其他经典非晶体体系也具有一定的特殊性。以聚合物聚对苯二甲酰对苯二胺[18]和铝酸三钙水泥[19]的分子动力学实验模拟结果为例进行对比,聚合物应力-应变曲线反映出典型的弹性形变-失效过程,水泥的分子动力学模拟图像随着应变发生出现明显的断裂层。由于弹性形变和塑性形变同时发生在沥青质聚集体中,且随着形变的发生2种形变的贡献也会发生变化,沥青质在实验范围内随应变上升不会出现明显的失效阈值。同时由于应变范围相对较小,难以从分子模拟图像上直接观察到材料整体受到破坏的现象。
1.3 沥青质形变势能变化
基于1.2节所述,对沥青质聚集体受力形变过程的描述应当从分子的形变和分子间相互作用的变化入手。分子动力学模拟基于分子构形或相互作用相关的标准参考值和实际值的差值与能量的拟合公式,对模拟系统的势能进行计算和统计,换言之,也可以通过势能反过来对系统内分子总体构形变化进行描述。势能项又可以分为分子内势能(由分子构型决定)和分子间势能(分子间相互作用)。在最简单的力场中,分子内势能可以分为键伸缩(Bond stretching)、键角弯曲(Bending)和二面角扭矩(Torsion)3类,分子间势能是静电相互作用和范德华相互作用之和。力场计算还考虑多个分子构型参数之间的相互作用,设立交叉项,这些项是对分子内势能的修正,在数据处理中可以将这些项整合进3种基本分子内势能中。同时考虑到sp2杂化的平面构型维持因素,统计出偏离平面构型附加势能[20]。
在真实沥青中,上千种沥青质分子堆积成沥青质聚集体进而成相。笔者采用势能来研究具有不同稠环链接结构的沥青质分子在应变过程中分子结构与分子间相互作用的变化,为了突出强调分子结构的影响,在每一个动力学模型盒子中只放置一种模型分子。考虑到沥青的非晶态相态变化特点,在工作温度范围(250~353 K)内可能会出现玻璃态、高弹态甚至到黏流态的相态变化过程,故每种模型分子模拟250、293和353 K 3个温度下的相态。为了模拟沥青质在正常工作条件和极大形变情况下的势能变化,应变分别选择±0.01、±0.03、±0.05和±0.10共计8个应变值。
含有不同沥青质的盒子在相同温度下总势能不同,含有同样分子的盒子在不同温度下总势能也不相同,因此比较不同分子或温度下盒子在形变过程中势能的变化值并没有太多意义。定义相对势能变化值(Relative potential energy change),即应变发生后系统势能的变化值与应变前系统势能的比值,来描述势能随应变发生的变化,从而可以描述沥青分子构型和分子间相互作用随应变发生的变化规律,也能直观比较相同应变下不同模型分子或温度系统的分子构型和分子间相互作用的变化情况。
2 实验条件
采用Material Studio 2017R2进行分子动力学模拟。软件支持无定形结构材料性质及相关过程的研究,笔者采用其中的Forcite模块对沥青质分子形成的介观微结构进行模拟。利用预设力场参数,通过有限差分法,采用数值积分求解所构建体系中的牛顿运动方程,从而获取每个分子随时间变化的坐标、构象、瞬时势能和动能,依据统计力学计算体系的整体性质。
实验力场采用COMPASS Ⅱ[21],相互作用截断半径为1.55 nm。动力学模拟分为模型构建和应变施加2个阶段。
通过模型构建阶段完成设定条件下沥青质聚集体的模拟。在无定形盒子中分别放入100个相同的沥青质模型分子作为沥青质聚集体模型,3种沥青质模型分子分别对应生成的3个盒子。模型构建阶段对每个盒子进行如下流程的动力学模拟:①对盒子进行能量优化;②对盒子进行250~500 K、5次循环的退火过程;③在1 GPa、NPT系综内对盒子进行体积收缩,每种沥青质模型盒子分别固定在250、293和353 K温度进行模拟,整个过程花费500 ps,步长1 fs,选择Nosé-Hoover恒温器和Andersen恒压器;之后分别在相同温度、正则系综下进行200 ps、步长0.5 fs的动力学模拟以维持系统稳定,采用Nosé-Hoover恒温器;此步完成时总计生成9个模型盒子;④各模型盒子的温度设定与第3步中相同,在1×10-4GPa、NPT系综内生成沥青质聚集体模型,总模拟时间5 ns,步长1 fs,选择Nosé-Hoover恒温器和Andersen恒压器;然后在相同温度、正则系综下进行200 ps、步长0.5 fs的动力学模拟稳定系统,采用Nosé-Hoover恒温器。由此获得沥青质聚集体的模型。
应变施加阶段中,对上一阶段优化好的9个模型分别施加±0.01、±0.03、±0.05和±0.1的应变,施加应变后合计72个模型盒子。应变的施加在与各自上一阶段相同的温度、正则系综下完成,整个过程花费500 ps,步长1 fs,选择Nosé-Hoover恒温器。
在势能项的读取中,将交叉项整合进键伸缩、键角弯曲和二面角扭矩3类势能项中。将伸缩-伸缩交差势整合到键伸缩势能项,将伸缩-弯曲-伸缩交叉势、弯曲-弯曲交叉势和扭矩-弯曲-弯曲交叉势整合到键角弯曲势能项,将伸缩-扭矩交叉势、伸缩-扭矩-伸缩交叉势和弯曲-扭矩-弯曲交叉势整合到二面角扭矩势能项。
3 实验结果
3.1 总势能变化
图2是3种模型分子As1、As2和As3在不同温度下体系相对势能变化与应变的关系。由图2可见,3种模型分子聚集体的正应变和负应变的势能变化图像并不对称,可见沥青质聚集体的应变不能用简单的弹性形变来描述。当应变超过±0.05时,体系的总势能常能观察到异常的变化,显示为剧烈的上升或异常下降。为了避免应变中材料性能变化或内部结构破坏,应该保证体系内势能在一定范围内平稳变化,沥青的设计应变不超过±0.03的经验和模拟计算的结果相吻合。因此对相对势能变化曲线应重点关注±0.03应变区间。
图2中,在±0.03应变范围内,250 K下3种分子聚集体模型的相对势能变化范围和趋势相近,显示出低温下势能相对变化和应变在一定范围内的关系与分子构型无关。在293 K下,As2和As3模型的势能变化曲线在±0.03应变范围与各自在250 K下的类似,但As1模型则在293 K下相对势能变化非常显著,且随应变绝对值上升而迅速上升。在353 K下,As1和As2的相对势能变化曲线较为接近,与各自在293 K下的势能变化曲线差别明显;而As3在353 K下相对势能变化曲线与250 K及293 K下的差别依旧不大。

图2 沥青质分子聚集体模型相对势能变化与应变的关系Fig.2 Relationships between strains and relative potential energy changes for asphaltene aggregation models(a) As1—Totally cata-condensed asphaltene; (b) As2—Partly cata-condensed asphaltene; (c) As3—Totally peri-condensed asphaltene
综上所述,沥青质的稠环缩合方式不同,在不同的温度下其聚集体随应变势能上升的趋势也有所不同。在±0.03应变范围内,迫位缩合的沥青质聚集体随应变势能上升最不明显,且温度对曲线的影响不显著;渺位缩合的沥青质聚集体随应变大幅上升,曲线形状和趋势受温度影响很大;而局部渺位缩合的分子在较低温度下与迫位缩合分子的曲线相近,高温(353 K)下则和渺位缩合分子的曲线接近。
3.2 势能变化类型分析
将沥青质聚集体模型在应变中相对势能变化分解为相对分子内势能变化和相对分子间作用变化,前者也可以称为相对形变势能变化(Relative deformation potential energy change)。对各模型中2种相对势能变化值和应变的关系进行分析。
图3是3种沥青质组成的模型在不同温度下相对形变势能变化值和应变的关系。由图3可知,3种分子组成的模型相对形变势能随应变变化的大小和规律各不相同,渺位缩合的As1组成的模型相对形变势能变化最大,As2次之,迫位缩合的As3相对形变势能变化最小。相对形变势能变化曲线的变化规律可以概括为2类:第一种是相对形变势能随应变绝对值增大而增加,无论是拉伸还是压缩模型形变势能均高于原始状态,曲线呈现U字形趋势;第二种是相对形变势能随应变增大而减小,即分子形变势能随压缩形变增大而升高,随拉伸形变增大而下降。也有一些形变势能变化曲线是2种规律叠加的结果。
图3中,在±0.01应变范围即沥青正常工作区内,293 K及以上温度的As1模型、所有温度下的As2模型和250 K时的As3模型均表现为U形趋势,其余则表现为一致下降的趋势。而在±0.03应变即正常工作应变边界时,As1在3个温度下的变化趋势均与±0.01时的趋势维持一致,As2在250 K 和293 K温度下0.01~0.03区间内相对势能曲线开始下降,而As3模型在250 K和293 K下在0.01~0.03应变区间也开始下降,353 K下却开始回升。总结来看,3种分子的模型的相对形变势能变化曲线和温度有如下关系:当温度较低(250 K)时,整体趋势倾向于一致下降的趋势;而在较高温度下(353 K),相对形变势能变化曲线均往U形趋势变化。

图3 沥青质分子聚集体模型相对形变势能变化值与应变的关系Fig.3 Relationships between strains and relative deformation potential energy changes for asphaltene aggregation models(a) As1—Totally cata-condensed asphaltene; (b) As2—Partly cata-condensed asphaltene; (c) As3—Totally peri-condensed asphaltene
图4是3种模型分子组成的聚集体模型的相对分子间相互作用势能变化值与应变的关系。如公式(1)所示,无应变和应变状态下分子间相互作用势能均为负值,因此若发生应变后分子间相互作用势能上升即分子间相互作用势能变化值为正,此时相对分子间相互作用势能变化为负值;且应变后势能上升越多,相对分子间相互作用势能变化值越小(该值的绝对值此时会上升)。同理,若发生应变后分子间相互作用势能下降,此时分子间相互作用势能变化值为负,相对分子间相互作用势能变化为正值;且应变后势能下降越多,则相对分子间相互作用势能变化值越大。

图4 沥青质分子聚集体模型相对分子间相互作用势能变化值与应变的关系Fig.4 Relationships between strains and relative intermolecular potential energy changes for asphaltene aggregation models(a) As1—Totally cata-condensed asphaltene; (b) As2—Partly cata-condensed asphaltene; (c) As3—Totally peri-condensed asphaltene

(1)
分子间相互作用势能变化=应变下分子间
相互作用势-无应变分子间相互作用势能
(2)
在分子动力学中,两分子间的相互作用势能遵循L-J势能曲线。从图4可以观察到,一些曲线(例见图4(c)中3条相对势能曲线)呈现倒U形,表明分子间相互作用势能随应变的变化和L-J势能曲线规律相似,倒U形的顶点对应的是分子间相互作用势能的最低点。另外有一些相对势能变化曲线单调递减(见图4(a)中293 K与353 K的相对势能曲线),这些案例意味着整个体系的相对分子间相互作用势能最低点在应变范围之外,随着应变由负到正逐渐增大,体系分子间相互作用势能逐渐增大。而在±0.03应变区间,所有的相对分子间相互作用势能变化值均随应变增大而下降,意味着在这一范围内分子间相互作用势能随应变增大而单调递增。
综合来看,沥青质聚集体模型随应变绝对值增大势能上升,是分子间相互作用势能和分子构形势能变化的共同结果,且在正应变(拉伸)和负应变(压缩)下影响不同。随着模型压缩程度上升,负应变绝对值逐渐增大,分子相对形变势能上升(见图3),相对分子间相互作用势能可能略微下降,但不会抵消总势能的上升趋势。而随着模型拉伸,正应变逐渐增大,分子相对形变势能在不同的模型中上升或先升后降,而相对分子间相互作用势能大幅上升,使总势能依旧上升。
3.3 形变势能类型分析
图5是3种分子在250、293和353 K下形成的9个聚集体模型中的伸缩、弯曲、扭矩和偏离平面构型附加4项势能变化。由图5可知,各项形变势能均处于规则不明显的波动状态,难以用简单的规律进行描述。尽管如图3所示,压缩过程必然增加分子形变势能,但依旧能从图5中观察到一些势能变化值小于0的点。由此可见模型整体的压缩并不会同步增大所有的分子形变势能项,分子形变势能的变化是多种形变形式共同协调作用的结果。

As1—Totally cata-condensed asphaltene;As2—Partly cata-condensed asphaltene;As3—Totally peri-condensed asphaltene图5 As1、As2和As3分子聚集体模型在250、293和353 K各类形变势能变化值与应变的关系Fig.5 Relationships between strains and several deformation potential energy changes of As1, As2 andAs3 models at 250, 293 and 353 K(a) Stretching; (b) Bending; (c) Torsion; (d) Out-of-plane
从伸缩势能项可以观察到,压缩形变时As3模型中该项的变化十分可观,而As2维持在一个较低的变化水平;拉伸形变时则正好相反,As2拉伸形变势能变化水平上升,As3的变化反而较低(见图5(a))。弯曲势能项变化值由高到低依次是As1、As2、As3,As3分子一直保持在很低的弯曲势能变化水平上,可见迫位缩合程度越高的沥青质分子键角在应变中变化越小(见图5(b))。3种分子模型的扭矩势能变化值显示,As3模型在应变中的扭矩变化远小于前两者(见图5(c))。综上所述,渺位缩合分子在聚集体应变过程中的形变主要以分子内部不同平面之间的夹角扭曲来实现,而不仅仅是将化学键进行拉伸、压缩或分子平面键角发生变化;迫位缩合的沥青质分子键角弯曲势能和二面角扭曲势能随应变的变化较小,分子整体呈现相对刚性,分子形变势能反而主要由键伸缩贡献。As1模型的3种形变势能变化项均最为明显,这可能是由渺位缩合的稠环链接方式和短饱和侧链2个因素共同所致。
沥青质的稠环芳核有维持平面构型的趋势。当整个模型被压缩时,分子构型会扭曲偏离平面构型,偏离平面附加势能会有一定程度的上升如图5(d)所示。所有的模型在不同温度下均符合随应变增大而下降的趋势。在压缩形变过程中,偏离平面构型附加势能在形变势能上升中起到不可忽视的作用。
4 结 论
(1)所有沥青质聚集体模型随应变的势能变化基本呈现U字形趋势。在沥青工作的±0.03应变范围内,全迫位缩合沥青质聚集体模型的相对势能变化值最小,全渺位缩合分子聚集体则最大。这意味着全迫位缩合的分子在近似弹性响应的状态下材料整体更容易发生形变,在设计形变范围内强度最低;而全渺位缩合分子正好相反。
(2)迫位缩合沥青质聚集体的相对势能变化值随应变的变化趋势受温度影响最小,渺位缩合分子聚集体受影响则相对更大。由此可见,迫位缩合沥青质分子尽管对材料整体强度贡献较低,但能够降低材料整体的感温性,保证在工作温度范围材料强度性能变化幅度较小。
(3)沥青质聚集体模型在应变下的势能增加主要由形变势能和分子间相互作用势能贡献,两类势能随应变变化的趋势各有特点。分子间相互作用势能在±0.03应变范围内随应变增加单调上升;分子形变势能则存在U字形或单调递减2种可能的趋势,同一种分子组成的模型在不同温度下变化趋势也可能不同。这意味着沥青质分子在材料拉伸和压缩状态下的贡献是不同的,在一些场合下,材料的形变也可能会使分子平均构型反而更接近能量最低的状态。
(4)不同构型的沥青质分子在聚集体应变中的分子形变类型也有所不同。渺位缩合分子主要依靠分子扭矩的变化提高分子形变势能。迫位缩合分子在压缩过程中则主要由于键伸缩带来势能变化,分子扭矩和键角弯曲对势能的影响均不明显。此外,由于沥青质分子均有尽力维持平面构型的稠合芳环结构,因此所有分子模型的平面维持附加势能均随着应变上升而下降。由此可见,沥青材料形变时沥青质分子立体构型提供了可观的内应力。
猜你喜欢
食品工业科技(2022年23期)2022-12-06
——《势能》
文化纵横(2022年3期)2022-09-07
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
军民两用技术与产品(2022年1期)2022-06-01
河南工业大学学报(自然科学版)(2021年6期)2022-01-26
化工科技(2021年5期)2021-11-24
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中国典型病例大全(2020年6期)2020-10-21
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
