
Ni2P催化剂电子结构对4,6-DMDBT加氢脱硫活性和选择性的影响
2022-07-19 03:48赵加民刘清河刘诗哲戴立顺
石油学报(石油加工) 2022年4期
韩 伟, 赵加民, 张 乐, 刘清河, 刘诗哲, 戴立顺, 袁 迎
(1.中国石化 石油化工科学研究院,北京 100083;2.聊城大学 化学化工学院,山东 聊城 252000)
With the growing severe environmental problems and increasing supply of high-sulfur crude oil, the hydrodesulfurization (HDS) technologies for producing ultra-low sulfur fuels have been facing more and more challenges[1-2]. As the general core role in HDS technologies, high-performance HDS catalysts have always been the driving force to meet these challenges. The current commercial HDS catalysts are generally constructed using Ni or Co-promoted MoS2nano-slabs as the active phases andγ-alumina (γ-Al2O3) or modified ones as the support[3-5]. However, due to the anisotropic structure of MoS2crystallites, the active sites are restricted to the edge sites of the MoS2and the majority of sites on the basal plane are inactive[6-8]. In very contrast to MoS2, the transition metal phosphide (TMP) catalyst has an isotropic structure, which is able to expose metal sites on all surface planes, thus resulting in a higher density of active sites and thereof a higher HDS activity than the above commercial sulfide catalysts[9-12]. Among various phosphides, Ni2P has the highest HDS activity, and shows about two-fold intrinsic activity relative to Co/Mo sulfide catalyst[10]. Moreover, Ni2P catalysts have recently been viewed as a tremendous potential candidate material for a new class of commercial HDS catalysts[13-14]. However, compared with the commercial Co(Ni)Mo sulfide catalysts, the excessively high hydrogen consumption of Ni2P-based catalysts during the HDS process due to their lower direct desulfurization (DDS) selectivity seriously limits their industrial application[13]. Therefore, how to overcome this limitation has become a most fundamental conundrum in the HDS chemistry of Ni2P-based catalysts.
The current prevailing theory about tuning HDS selectivity of Ni2P catalyst is mainly proposed by Oyama group[10,15-17]. They proposed that the DDS selectivity is controlled by the number of tetrahedral Ni(1) sites in Ni2P phase, while the square pyramidal Ni(2) sites are considered as the high-activity sites for hydrogenation desulfurization (HYD) pathway. The mechanism was also applied to account for the superior DDS selectivity of Ni-Fe-P systems as compared with Ni2P catalysts[18-19]. The unprecedented DDS selectivity of Ni-Fe-P was attributed to the reconstruction of the Ni-Fe phase which exposed more Ni(1) sites. However, the DDS selectivity (≈70%) in Ni/Fe/P/SiO2(The molar ratio of Ni to Fe is 3∶1) largely overlapped the amount of Ni(1) sites (≈45%). This observation indicates that the HDS activity of Ni(1) sites is probably much more active than that of Ni(2) sites, however, it is contrary to the previous research[20]. According to these studies, besides the crystal phase structure, the HDS activity and selectivity of Ni2P could be influenced by other important factors.
Apart from the crystal phase structure, electronic properties are also the essential factor to decide the HDS performance of Ni2P-based catalytic systems[13]. It is demonstrated that the electron density around the metal had a good correlation with HDS activity of thiophene with the order that Ni2P/SiO2>MoP/SiO2>MoS2/SiO2[21]. Meanwhile, the importance of electronic perturbations of Ni2P on HDS selectivity was manifested recently by Yong-Kul Lee and coworkers[13], in which Ni2P nanoparticles contacting with Ga-SiO2support showed enhanced electron deficient Ni2P than Ni2P/SiO2and finally favoredσ-bonding with S compounds to promote direct desulfurization of 4,6-DMDBT. However, so far it seems very difficult to accurately understand the electronic effects due to the great challenge of fabricating model Ni2P catalysts with tunable electronic structure meanwhile maintaining the similar crystalline structure. In other words, before carrying out a deep insight into the relations between the electronic structure of Ni2P-based catalysts and their HDS activity and selectivity, a series of model catalysts should be fabricated.
However, since the traditional hydrogen temperature-programmed reduction (H2-TPR) synthesis methods of metal-doped Ni2P/SiO2always caused the formation of alloy phases due to the utilization of high temperature (Generally from 500—700 ℃), the crystalline structure of the resulting Ni2P-based catalysts are often unavoidably changed[15,19,22]. Herein, to avoid this phenomenon to accurately study the relations between the electronic structure and the HDS functions of Ni2P-based catalysts, a low-temperature synthesis method of metal phosphides was introduced to prepare the metal-doped Ni2P catalysts, and the preparation procedure is shown in Fig.1. Firstly, silica-supported nickel phosphide (Ni2P) was synthesized by the H2-TPR method at a high temperature of 650 ℃. Secondly, the resulting undoped Ni2P/SiO2catalyst were divided into three parts and doped by Co, Fe and Mo by in-situ phosphitylation method at a low temperature of 300 ℃, respectively. Herein, for effectively tuning the electronic properties of undoped Ni2P/SiO2catalyst, the selection basis for these three metals mainly depends on their different or similar electronegativity, i.e. Mo (2.16), Co (1.88), and Fe (1.83). Fourier transform infrared (FTIR) spectroscopy using CO as the probe molecule was used to gain insight into the nature of the surface-active sites. The HDS activities were assessed using the most refractory 4,6-DMDBT as the model reactant. Results clearly show that the electronic structure of Ni2P was successfully tuned by introducing the three metals without formation of bimetallic alloying phases of phosphides, thus laying a foundation to effectively study the electronic effects of Ni2P active phase on the HDS activity and selectivity.
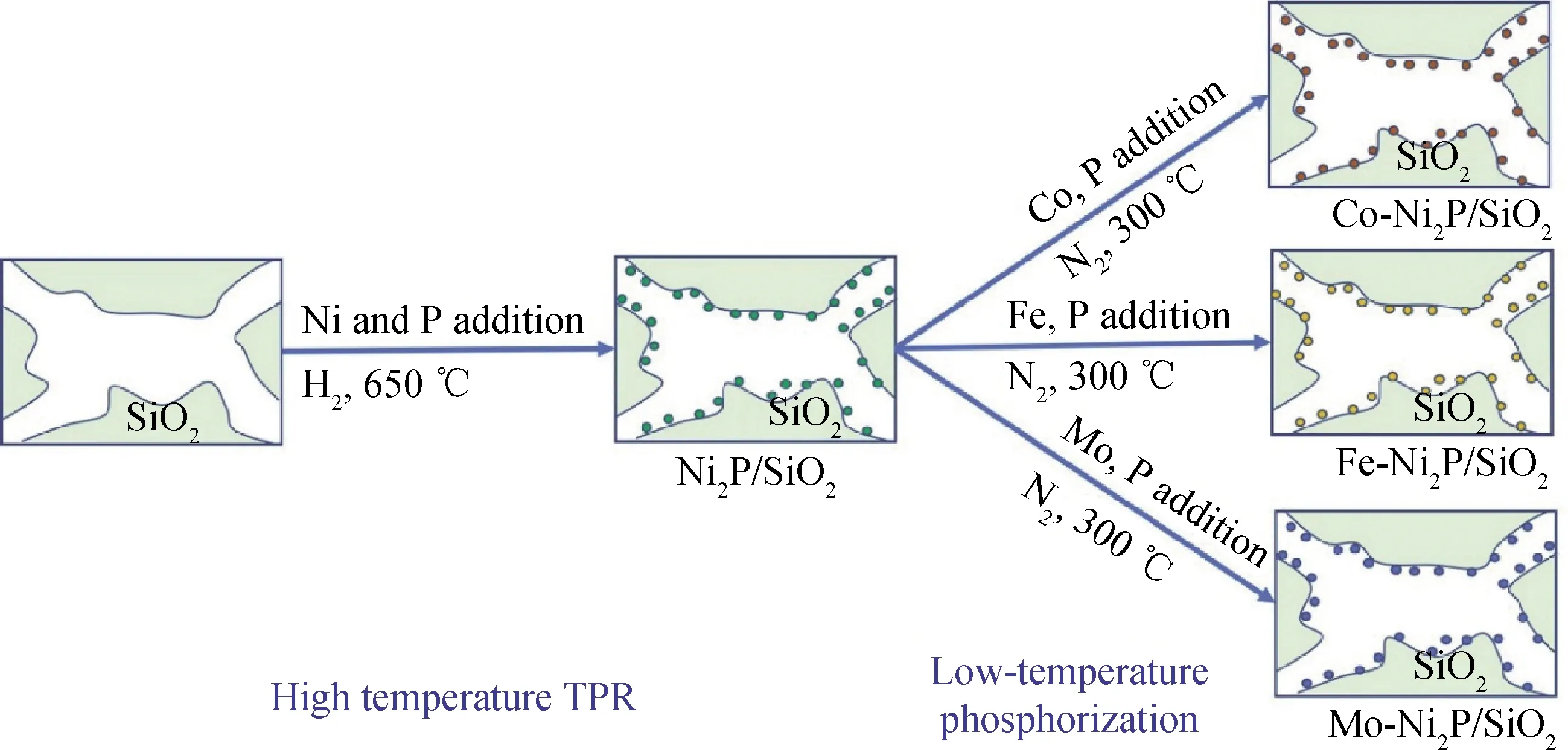
Fig.1 Fabrication processes of Fe-, Co- and Mo-doped silica-supported Ni2P catalysts
1 Experiment
1.1 Raw material and reagent
Silica (SiO2, Cab-O-Sil, M-7D grade, 150 m2/g), nickel nitrate hexahydrate (Ni(NO3)2·6H2O, Alfa, 98%), ammonium phosphate ((NH4)2HPO4, Samchun, 99%), nitric acid, ferric nitrate nonahydrate (Fe(NO3)2·9H2O, Alfa, 98%), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, Alfa, 99%), and Ammonium heptamolybdate tetrahydrate ((NH4)2Mo2O7·4H2O, Alfa, 98%). All chemical reagents were purchased from Innochem and used without further purification.
1.2 Synthesis of silica-supported Ni2P and three doped Ni2P catalysts
The Ni2P/SiO2catalyst was prepared by incipient wetness impregnation of the silica support (SiO2, Cab-O-Sil) with dissolved nickel nitrate hexahydrate (Ni(NO3)2·6H2O), ammonium phosphate ((NH4)2HPO4) and several drops of nitric acid in distilled water. The Ni loading amount was fixed at 1.16 mmol Ni per 1 g support by controlling the initial Ni/P molar ratio of 1.0. After impregnation, the catalyst sample was dried at 120 ℃ for 12 h, calcined at 500 ℃ for 6 h, and then cooled to room temperature. The oxidic sample was reduced in a quartz U-tube reactor by temperature-programmed reduction (TPR) from room temperature to 650 ℃ at 5 ℃/min and held at 650 ℃ for 2 h in a 10%H2/Ar molar flow (100 cm3/min). After TPR, the catalyst sample was cooled to room temperature and passivated at 50 cm3/min of 0.5%O2/Ar molar flow for 4 h.
Because the procedures for preparing Fe/Co/Mo-doped Ni2P/SiO2catalysts were similar, only the preparation procedure Fe-doped Ni2P/SiO2(denoted as Fe-Ni2P/SiO2) was given here. Firstly, undoped Ni2P/SiO2was prepared according to the above H2-TPR methods. Secondly, the aqueous ferric nitrate nonahydrate (Fe(NO3)2·9H2O, Alfa, 98%) solutions were prepared and then used to impregnate undoped Ni2P/SiO2by incipient wetness impregnation method. The amount of Fe loading was fixed at the molar ratio of Fe/(Ni+Fe) is 0.3. The molar ratio is the optimal for electronic effects in conventional HDS sulfide catalysts, which found in our previous group studies[5,23-24]. After impregnation, the catalyst sample was dried at 120 ℃ for 12 h. Then, the in-situ phosphitylation method was used to prepare Fe-Ni2P/SiO2. Typically, the stoichiometric amount of sodium hypophosphite (P/(Ni+Fe) molar ratio of 4) is placed at the upstream side of the tube furnace and the correspond precursors are placed at the downstream side. The samples are heated at 300 ℃ for 120 min with N2gas flowing at 30 mL/min. Finally, the catalyst sample was cooled to room temperature and passivated at 50 cm3/min of 0.5%O2/Ar molar flow for 4 h. Similarly, the Co-Ni2P/SiO2and Mo-Ni2P/SiO2was prepared with the Co(NO3)2·6H2O and (NH4)2Mo2O7·4H2O, respectively.
1.3 Catalysts characterization
X-ray diffraction (XRD) measurement was used to analyze the crystalline structures of the different catalysts, which performed on a Bruker D5005 X′Pert diffractometer with CuKαradiation (λ=0.15406 nm) at 60 kV and 300 mA. The 2θrange was between 5° and 80° and the scanning speed was 2 ℃/min.
The surface compositions of the different catalysts were analyzed by XPS measurement with an ESCALab250 electron spectrometer (Thermo Fisher Scientific Corp) with monochromatic 150 W AlKαradiation. The pass energy for the narrow scan of 30 eV and base pressure of 6.5×10-8Pa was chosen. Quantification of surface content of elements was done using sensitivity factors provided with VG software. Ni species′, P species′, Fe species′, Co species′ and Mo species′ chemical states and their contents can be distinguished and quantified by XPS spectrum fitted by XPSPEAK software (Version 4.1).
Surface areas and average pore diameters of the different catalysts were characterized by Micromeritics ASAP 2002 micropore size analyzer from the linear portion of BET plots (p/p0=0.01—0.10) at liquid nitrogen temperature. Prior to the experiment, the sample was degassed at 200 ℃ for 15 h under a vacuum of 0.00133 Pa, and then cooled to room temperature. The surface area was determined according to the BET method and the pore volumes were obtained from N2adsorption-desorption isotherms.
The morphology and structure of the catalysts were characterized by HRTEM on a Philips Tecnai G2 F20 instrument, operated at a primary electron energy of 220 keV, a point-to-point resolution of 0.19 nm. The images were performed on a TVIPS 1k×1k CCD camera. Elemental mapping images were collected using a TEM equipped with EDX spectroscopy. The catalyst samples for STEM were first ultrasonically dispersed in ethanol, and then the testing samples were prepared by dropping the dispersed suspensions samples onto carbon-coated copper grids.
Infrared spectra of CO chemisorption at low temperature were obtained for the passivated catalyst samples (≈50 mg) in a reactor cell placed in Perkin Elmer Frontier FTIR spectrometer at 0.25 s data scan interval with a resolution of 1 cm-1. The samples were pressed into discs and placed in a quartz IR cell with water-cooled KBr windows. Before dosing CO, the catalyst samples were reduced in 100 cm3/min H2flow at 450 ℃ for 2 h under the same conditions used for HDS reactivity studies, then cooled to room temperature in a N2flow, and exposed to CO flow until saturation was achieved.
According to the Beer-Lambert law, the concentration of site (n, μmol/g) on which CO is adsorbed can be calculated by Equation (1):
n=(A·S)/(ε·m)
(1)
whereεis the associated CO molar extinction coefficient (cm/μmol),Ais theνCO(Infrared wave number of CO) band area (cm-1),Sis the catalyst′s surface (cm2), andmis the mass of the catalysts (g). Based on the researches of Oliviero′s groups[25], when theνCOat about 2090 cm-1, the CO molar extinction coefficient is about (20±3) cm/μmol. Thus, the turnover frequency (TOF) of the HDS reaction can be calculated by Equation (2):
TOF=-F·ln(1-X)/(m·n)
(2)
where,Fis the molar amount of 4,6-DMDBT fed to the reactor per second (μmol/s),Xis the conversion of 4,6-DMDBT (%),mis the catalyst mass (g) andnis molar concentration of site (μmol/g). The details of TOF of the HDS reaction are listed in Table 1.

Table 1 CO adsorption on the three doped catalysts and the undoped Ni2P/SiO2 catalyst
1.4 Catalytic performance assessment
The HDS performance of the three doped Ni2P/SiO2catalysts (Fe-Ni2P/SiO2, Co-Ni2P/SiO2and Mo-Ni2P/SiO2), and the monometallic (Ni2P/SiO2) catalysts were carried in a fixed microreactor which has been described in detail in our previous studies[23,26]. HDS activity measurements were carried out using a reactor feed consisting of mass fraction 0.45% 4,6-DMDBT (Provided by J & K Chemical Company) inn-decane. A sample of 0.20 g catalyst (40—60 mesh) was diluted with 1.0 g 40—60 mesh quartz sand before being loaded into the reactor. Mass transport and heat calculations were carried out in our previous studies[26-27]. It is demonstrated that the particle size of 40—60 mesh can effectively avoid the mass and heat transfer limitations.
Before the HDS test, the catalysts were all treated at 450 ℃ in 360 cm3/min H2flow for 2 h under a hydrogen pressure of 4.0 MPa in the reactor. After reduction, the temperature decreased to the reaction temperatures of 280, 300, 320 and 340 ℃. The LHSV of the HDS tests is 58.4 h-1, and the H2/feed volume ratio is 500. In each HDS reaction process, a reaction stabilization state would be reached after 2 h at various reactions temperatures. During the stabilization period, the composition and distribution of the product keeps constant, which thus could be used to check the deactivation during testing. Then the reaction products were analyzed with Agilent 7890A gas chromatograph equipped with a flame ionization detector (FID).
2 Result and discussion
2.1 Characterization results of silica-supported Ni2P catalysts
2.1.1 XRD
Fig.2 shows the XRD patterns of Ni2P/SiO2and its Fe-, Co- and Mo-doped counterparts. In the XRD pattern of Ni2P/SiO2, the sharp diffraction peaks at 40.5°, 44.3°, 47.1° and 54.0° can be attributed to the (111), (201), (210) and (300) planes of Ni2P, respectively, which confirms that Ni2P particles were synthesized successfully[15,28-29]. The Fe-, Co- and Mo-doped Ni2P catalyst samples show almost the same XRD patterns as Ni2P/SiO2, indicating that Ni2P phase dominated in all these samples. There is no extra peak of bimetallic catalysts in Fe-, Co- and Mo-doped Ni2P catalyst samples, which indicates that the bimetallic alloy phase of phosphides (Generally, NiFeP, NiCoP, NiMoP) has hardly been reconstructed. Besides, the characteristic peaks of CoP, FeP and MoP cannot be detected in these doped catalysts. Considering that the detectability of XRD analysis is about 4 nm, the above results imply that the uniform distribution of CoP, FeP and MoP phase into Ni2P/SiO2[30].

Fig.2 XRD patterns of Ni2P/SiO2, Co-Ni2P/SiO2, Fe-Ni2P/SiO2 and Mo-Ni2P/SiO2
2.1.2 HRTEM
To characterize the change of the nano-structure of Ni2P phases in different catalysts, the HRTEM images of the four catalysts were taken and the representative ones are shown in Fig.3. The TEM image of the four catalysts only showed the amorphous silica form, in which the spherical Ni2P particles are well dispersed on the SiO2. Moreover, the slice of the four catalysts was selected for HRTEM measurement to determine the interplanar spacings. As seen in Fig.3, the interplanar spacing for Ni2P is about 0.22, corresponding to the (111) planes, which matches well with the XRD data shown in Fig.2. In the three doped catalysts, there is also found the corresponding interplanar spacing for CoP, FeP and MoP, which demonstrated that the low temperature phosphitylation method can synthesize homogeneous mixtures, i.e. Ni2P-CoP, Ni2P-FeP, Ni2P-MoP, rather than bimetallic alloying phases. Furthermore, energy dispersive X-ray (EDX) was performed to understand the distribution of Ni species and other metal species in the synthesized doped catalysts, and the representative images of the sample Co-Ni2P/SiO2are shown in Fig.4. Clearly, the particles are confirmed to contain Ni, Co, and P, indicating a good mixture of the three elements (Fig.4 (b)—(e)).

Fig.3 HRTEM images of the catalysts(a1), (a2) Ni2P/SiO2; (b1), (b2) Co-Ni2P/SiO2; (c1), (c2) Fe-Ni2P/SiO2; (d1), (d2) Mo-Ni2P/SiO2
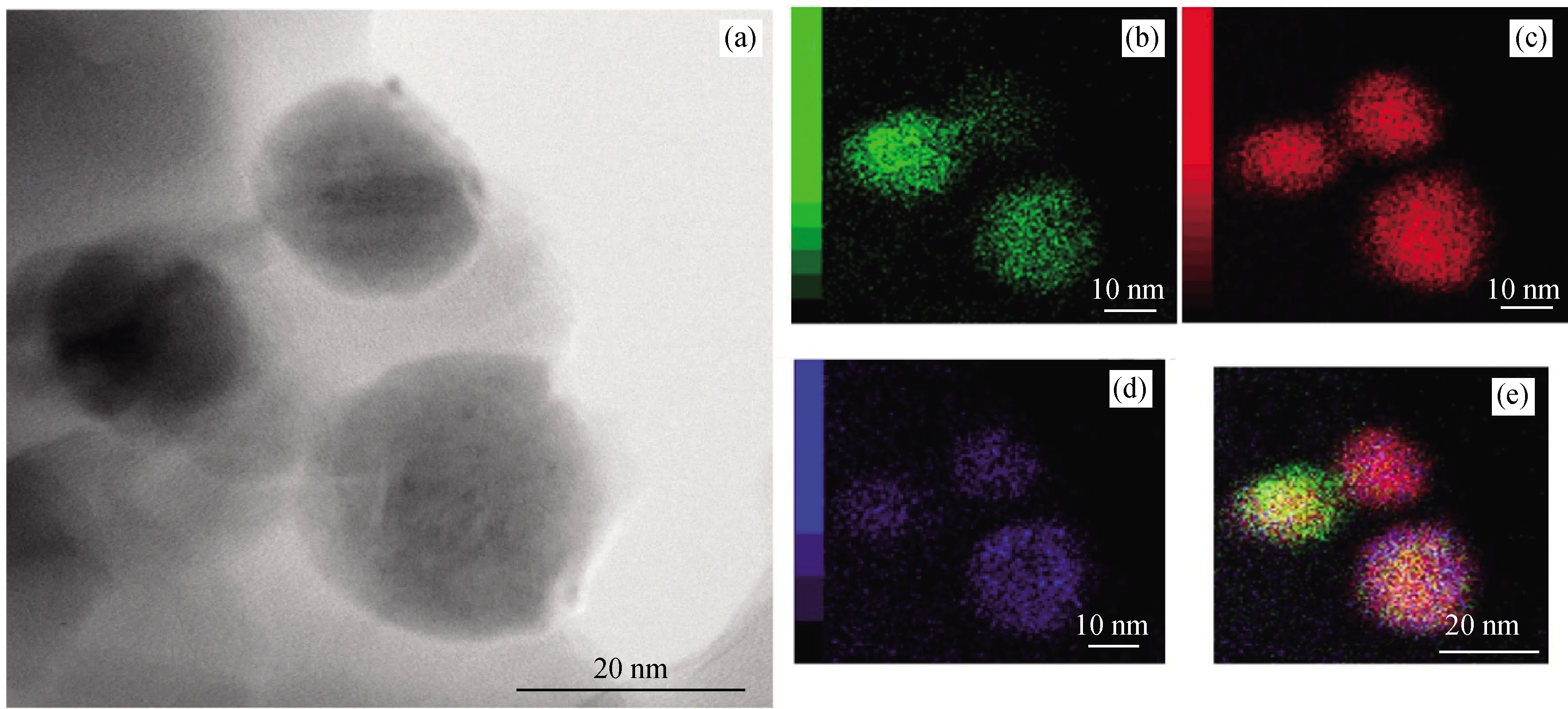
Fig.4 EDX images for Co-Ni2P/SiO2(a) TEM image; (b)—(e) Element mapping images of Ni (red), Co (green) and P (blue) show the element distributions.
2.1.3 XPS
The surface chemical compositions of the silica-supported TMPs catalysts were then investigated by XPS and the Ni 2p, Fe 2p, Co 2p, Mo 3d and P 2p spectra were shown in Fig.5. The binding energies of the Ni 2p3/2peak (Fig.5(a)) for Ni-P appear at about 853.10 eV, while those peaks at 856.50 and 861.76 eV arise from oxidized nickel species in the passivation layer formed on the Ni2P particles[31-32]. The P core level peaks (Fig.5(e)) are located at 129.5 eV, which corresponds to P 2p3/2of Ni2P phases. In the XPS spectra of P 2p, peaks at 133.7 and 135.1 eV correspond to the oxidized states 2p3/2and 2p1/2, respectively, which also originate from the surface oxidation. Furthermore, the binding energies of Ni 2p centered at 853.10 eV is positively shifted from the position of elemental Ni (852.5—852.9 eV)[33], and that of P 2p centered at 129.5 eV is negatively shifted from the position of elemental P (129.9 eV)[34], which implies that Ni carries a partial positive charge (δ+), where 0 <δ< 1, whereas the P has a partial negative charge (δ-), where 0<δ<2.
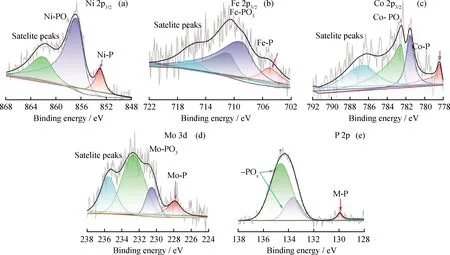
Fig.5 XPS survey spectra of the measured and curve-fitted Ni 2p3/2, Fe 2p, Co 2p and Mo 3d orbits of catalysts(a) and (e) Ni2P/SiO2; (b) Fe-Ni2P/SiO2; (c) Co-Ni2P/SiO2; (d) Mo-Ni2P/SiO2
In the Fe 2p spectra (Fig.5(b)) of Fe-Ni2P/SiO2, the binding energy peaks located at 706.55 eV are assigned to the Fe 2p states of Fe-P, and the peaks at 711.82 eV are assigned to its oxidized counterpart (Fe-POx)[35]. The high-resolution Co 2p3/2spectrum (Fig.5(c)) of Co-Ni2P/SiO2consists of component peaks at 778.37, 781.53 and 782.48 eV corresponding to CoP, oxidized cobalt species and cobalt hydroxide, respectively[31,36]. In the Mo 3d spectra (Fig.5(d)) of Mo-Ni2P/SiO2, the Mo 3d5/2peaks at 227.79, 230.45 and 232.60 eV are attributed to Moδ+in Mo-P, Mo4+and Mo6+ions, respectively[37-38]. The detection of these oxidized species could be attributed to the surface oxidation of phosphide in the passivation process. In addition, due to the results derived from XRD (Fig.2), the catalyst still manifests the well-crystallized Ni2P and does not indicate any discernible peaks of the oxides, which implies a mere surface-oxidation(less than 10 nm) that could only be detected by XPS[38]. From the XPS analysis, it could be concluded that (i) The outermost surface of catalysts is oxidized to phosphates, (ii) The detected Fe-P, Co-P and Mo-P peaks at 706.55, 778.37 and 227.79 eV indicate that successful synthesis of FeP, CoP and MoP on Ni2P/SiO2surfaces.
For characterizing the coordination environments and the electronic structures of Ni2P in the 4catalysts samples, the Ni 2p spectra in all catalysts were compared, as shown in Fig.6. Clearly, a shift for the binding energies of Ni-P of the doped catalysts in respect to the Ni2P/SiO2is observed as the following: A down shift of 0.09, 0.20 eV for Co-Ni2P/SiO2and Fe-Ni2P/SiO2catalysts, and a upshift of 0.13 eV for Mo-Ni2P/SiO2catalyst, which indicates that partial electrons are transferred from Fe, Co to Ni, and Ni to Mo. Those electron transfer is consistent with the Pauling′s electronegativity that Ni (1.91) is lower than Mo (2.16), and higher than Co (1.88) and Fe (1.83)[37]. These results suggest that the presence of Mo species around Ni2P could withdraw a part of electrons from Ni2P and reduce the electron density of Ni2P, while the presence of Fe and Co could increase the electron density of Ni2P[39].

Fig.6 XPS survey spectra of the measured and curve-fitted Ni 2p orbits of fresh Ni2P/SiO2, Co-Ni2P/SiO2, Fe-Ni2P/SiO2 and Mo-Ni2P/SiO2
2.1.4 Infrared spectroscopy of adsorbed CO
To provide deeper insight into the nature of electron deficient or rich of Ni2P phase on the three doped Ni2P/SiO2catalysts, infrared spectroscopy of adsorbed CO is carried out, which is always used as a probe for surface structure and chemistry of catalyst[13,40-41]. It is reported that the main vibrational banding for adsorbed CO species on Ni2P are observed in the wave number region of 2220—1900 cm-1, where four types of bonding are found: (1) CO adsorbed on unsaturated Niδ+(0<δ<1) sites at 2080—2099 cm-1, (2) CO terminally bonded to P (P=C=O species) between 2191 and 2186 cm-1, (3) Nickel tetracarbonyl formation (Ni(CO)4) around 2050 cm-1, and (4) CO adsorbed on Niδ+bridge sites at 1910 cm-1[40].
Fig.7 shows the CO adsorbed IR spectrum for the Ni2P/SiO2, Fe-Ni2P/SiO2, Co-Ni2P/SiO2and Mo-Ni2P/SiO2catalyst, which gives a distinctive IR band at 2082—2092 cm-1, which is attributed to the CO on atop Ni sties (cus Niδ+(0<δ<1) sites) on surface of these reduced catalysts. A previous study of CO chemisorption of Ni2P/Ga-SiO2has revealed that the shift ofνCObands to a higher frequencies than Ni2P/SiO2means the electron-deficient nature of Ni2P caused by electron-withdrawing Ga species[13]. In the present study, the order for frequency ofνCObands is observed: Fe-Ni2P/SiO2

Fig.7 Infrared spectra of adsorbed CO on reduced Ni2P/SiO2, Co-Ni2P/SiO2, Fe-Ni2P/SiO2 and Mo-Ni2P/SiO2
2.2 HDS performance
4,6-DMDBT has been widely considered as a typical representative of the unreactive sulfur compounds in transportation fuels[3,42], and herein it was chosen as the model reactant to assess the performance of HDS catalysts in the present study. The HDS activities of the three doped Ni2P/SiO2catalysts and the one undoped Ni2P/SiO2catalyst are shown in Table 2. The conversions of the undoped catalyst (Ni2P/SiO2) and the three doped (Fe-/Co-/Mo-) catalysts in our work are all below 50%, and only that of the Co-Ni2P/SiO2is higher than 40%. So, the activity assessment results could be used to rationally discuss the structure-function relations. Clearly, as shown in Table 2, as the same conditions, the HDS intrinsic activity of the four catalysts follow the clear-cut order of Co-Ni2P/SiO2>Ni2P/SiO2>Fe-Ni2P/SiO2>Mo-Ni2P/SiO2. The addition of Co was found to increase the HDS intrinsic activity, while the introduction of Fe and Mo resulted in a decrease of HDS intrinsic activity. Besides, with the reaction temperature increasing from 280 to 340 ℃, their intrinsic activity increment follows the similar order (Fig.8), which means that the more active catalyst, the more temperature-sensitive and the more obvious performance advantages[26].

Table 2 HDS results over the three doped Ni2P/SiO2 catalysts and Ni2P/SiO2 catalyst

Fig.8 HDS conversion of Ni2P/SiO2, Co-Ni2P/SiO2, Fe-Ni2P/SiO2 and Mo-Ni2P/SiO2Reaction conditions: H2 pressure 4.0 MPa; H2 volume flow 365 cm3/min; LHSV 58.4 h-1; H2/feed volume ratio 500
Reaction conditions: Temperature 280 ℃; H2pressure 4.0 MPa; H2volume flow 365 cm3/min; LHSV 58.4 h-1; H2/feed volume ratio 500; HYD/DDS—Selectivity ratio of hydrogenation desulfurization pathway/direct desulfurization pathway Considering that the surface areas and average pore diameter of the four catalyst samples are similar to each other (Table 3), and the Ni2P sites in the four catalysts have a tunable electronic structure meanwhile maintaining the similar crystalline structure, the electronic factors should be responsible for these variations in HDS catalytic activity. As observed in the present study[43-45], the higher electronic density on the active sites are expected to have high HDS reactivity, due to that the higher electron density of the metal atoms could improve the dissociation of H2and the adsorption to thiophenes. In fact, the effect of bonding interactions between metals and 4,6-DMDBT or its decomposition products on their HDS performance is more complex. Since the infrared spectroscopy of adsorbed CO could provide deeper insight into the nature of the bonding interactions between π bonds and metals surfaces[46], which is similar to the bonding between 4,6-DMDBT and metals surfaces[19,40], the IR band of CO vibration shifts showed in Fig.7 also indicate a change in strength of bonding interactions between 4,6-DMDBT and Mo-Ni2P/SiO2, Ni2P/SiO2, Co-Ni2P/SiO2and Fe-Ni2P/SiO2samples. The lower frequencies of CO vibration suggest stronger interactions between 4,6-DMDBT and metals surfaces. The effect of bonding interaction of π-metals on HDS performance was shown in Fig.9. There is a volcano-type relationship between CO vibration and HDS activity, meaning that a moderate interaction between active sites and 4,6-DMDBT is important for HDS intrinsic activity. For low bonding interaction, activation of 4,6-DMDBT is not sufficient for further reaction[47]. On the other hand, for high bonding interaction, activation of 4,6-DMDBT is possible, but the probability of deactivation of an active site by S adsorption will be higher. This relationship is explained by the basic idea that a medium value of electron density of Ni2P optimizes the appropriate interaction between sulfur organic compounds and active sites[48]. The present work provides the direct relationship between the interaction of reactants to active sites and HDS activity for transition metal phosphides.

Table 3 Physical properties of catalyst samples

DDS—Direct desulfurization; TOF—Turnover frequencyFig.9 HDS activity and DDS selectivity vs. CO vibration for Ni2P/SiO2, Co-Ni2P/SiO2, Fe-Ni2P/SiO2 and Mo-Ni2P/SiO2Reaction conditions: Temperature 280 ℃; H2 pressure 4.0 MPa; H2 volume flow 365 cm3/min; LHSV 58.4 h-1; H2/feed volume ratio 500
The HDS of 4,6-DMDBT mainly proceeds through two routes[49]: One is the so-called direct desulfurization (DDS) pathway to produce 3,3-dimethylbiphenyl (3,3-DMBP), and another is through the hydrogenation (HYD) pathway to obtain first 4,6-tetrahydro- and hexahydro-dibenzothiophenes, which are then hydrodesulfurized to methyl cyclohexyl toluene (MCHT) and 3,3-dimethylbicyclohexyls (3,3 DMBCH)[50]. These HDS products of 4,6-DMDBT were analyzed by a GC chromatograph, as shown in Fig.10. Clearly, the peaks at retention time 8—10.2 min were assigned to DMBCH while the peak at retention time of 10.2—12.1 min was assigned to DMCHB products. These DMBCH and DMCHB are the HYD products of 4,6-DMDBT. As compared, the peaks at 12.4—14.3 min were assigned to DMBP, which is the DDS products of 4,6-DMDBT. According to the results of GC chromatograph, the selectivity results of the two reaction pathways (HYD and DDS) over the three doped catalysts and Ni2P/SiO2catalyst are shown in Table 2. Using 4,6-DMDBT as the reactant, the HYD activity of Ni2P/SiO2was 16 times greater than the DDS activity. Similar results were also obtained over the three doped Ni2P/SiO2catalysts, with the selectivity of DDS product DMBP lower than 8% over Fe-Ni2P/SiO2, Co-Ni2P/SiO2and Mo-Ni2P/SiO2, indicating the 4,6-DMDBT HDS reactions mainly proceed through HYD pathway, which is consistent with other results[10,16-17]. The low selectivity to DDS is due to steric hindrance effects associated with the methyl groups of 4,6-DMDBT[3].

Fig.10 GC results of HDS products with Ni2P/SiO2, Co-Ni2P/SiO2, Fe-Ni2P/SiO2 and Mo-Ni2P/SiO2Reaction conditions: Temperature 320 ℃; H2 pressure 4.0 MPa; H2 volume flow 365 cm3/min; LHSV 58.4 h-1; H2/feed volume ratio 500
According to previous studies, theσ-bond adsorbing between S atom of 4,6-DMDBT and catalytic active sites is the key for DDS pathway, which leads to C—S bond cleavage[50-51]. Theσ-bond formation is facilitated particularly on the electron-deficient sites due to the lone electron pair of the S atom in 4,6-DMDBT. Thus, the electron deficiency of the Ni2P is beneficial for the DDS selectivity. On the other hand, higher electron density of the active center favors the formation of π-bond with the benzene ring of the benzo-thiophenic molecules, which is the key for HYD pathway (the C=C bond in benzene ring of the benzo-thiophenic is first saturated, and then followed by C—S bond cleavage)[13]. In the present research, the changes in HDS selectivity may be affected by the adjustment of the electron density of the catalyst caused by the doping of Co, Fe and Mo species, which changes the adsorption of S to the active site of Ni2P. According to the XPS characterization results (Fig.6), compared with undoped Ni2P/SiO2, Mo-Ni2P/SiO2has amore electron-deficiency in Ni2P, while Fe-Ni2P/SiO2and Co-Ni2P/SiO2has a higher electron density in Ni2P. Combined with the HDS evaluation results, it can be proposed that the increasing of the electron density of the Ni2P leads to higher selection for HYD during hydrodesulfurization of 4,6-DMDBT, which consists with the previous insight that higher electron density of active sites promotes to form the π-bond with 4,6-DMDBT and makes the C=C bond of aromatics to be firstly saturated following HYD pathway[13]. In contrast, the DDS pathway is mainly influenced by the surface electron deficient of Ni2P. Moreover, the XPS analysis of the spent catalysts was added for better understanding the electronic effect of catalysts during reaction, as seen in Fig.11. For Ni2P/SiO2(Fig.11(a)), the fresh sample shows a lower binding energy of Ni 2p3/2relative to the Ni-P peak, while the spent catalyst shows an increased Ni-P peak binding energy. The stronger Ni-P peak binding energy is consistent with disruption of metal-P and metal-metal bonds because of formation of a surface sulfide. In order to further analyze the electron effect of the doping metals, the binding energy of Ni 2p3/2of the spent doped catalysts was compared, as shown in Fig.11(b). Clearly, after HDS reaction, the binding energies of Ni 2p3/2still kept the similar order for those doped catalysts: Mo-Ni2P/SiO2>Ni2P/SiO2>Co-Ni2P/SiO2>Fe-Ni2P/SiO2, indicating the maintenance of partial electron transfer from Fe, Co to Ni, and Ni to Mo. Moreover, the CO-adsorbed FT-IR analysis (Fig.7) further confirmed that compared with undoped Ni2P/SiO2, Mo-Ni2P/SiO2with more electron-deficiency in Ni2P exhibits higher DDS selectivity, while Fe-Ni2P/SiO2and Co-Ni2P/SiO2with higher electron density in Ni2P have lower DDS selectivity. Thus, the fundamental research implies that modifying the active phases of Ni2P by an electron-inducing effect should be an effective strategy to improve the HDS selectivity, shedding a light on the rational design and development of high-efficient HDS catalysts. Based on this observation, a series of Al2O3supported TMPs was prepared by H2TPR methods, and their DDS selectivity is illustrated in Fig.12. The Ni2P/Al2O3and Fe-Ni2P/Al2O3had a superior DDS selectivity compared with the commercial CoMoS catalysts, which was accordingly attributed to the electron-deficiency of Ni2P sites caused by the stronger MSI between Al2O3and TMPs[52-53].

Fig.11 XPS survey spectra of the measured and curve-fitted Ni 2p orbits of spent Ni2P/SiO2, Co-Ni2P/SiO2, Fe-Ni2P/SiO2 and Mo-Ni2P/SiO2(a) The Ni 2p of fresh Ni2P/SiO2 and spent Ni2P/SiO2; (b) The Ni 2p of Mo-Ni2P/SiO2, Ni2P/SiO2, Co-Ni2P/SiO2 and Fe-Ni2P/SiO2

DDS—Direct desulfurization; HYD—Hydrogenation desulfurizationFig.12 HDS selectivity for Ni2P/SiO2, CoMoS/Al2O3, Fe-Ni2P/Al2O3 and Ni2P/Al2O3Reaction conditions: Temperature 340 ℃; H2 pressure 4.0 MPa; H2 volume flow 365 cm3/min; LHSV 58.4 h-1; H2/feed volume ratio 500
Overall, once the Ni2P phases are decorated, the electron cloud density of the neighboring Ni atoms will be changed, which will tune the interaction between active sites and 4,6-DMDBT, thereby changing the HDS activity and selectivity. The HDS occurred preferentially by HYD pathway for the Ni2P catalyst, and the HYD pathway was promoted by an increase of electron density of the Ni2P. DDS pathway is ascribed to the electron-deficiency of Ni2P sites.
3 Conclusion
In summary, silica-supported doped nickel phosphide (Ni2P/SiO2) with tunable electronic properties were synthesized by a two-step method: Firstly, silica-supported nickel phosphide (Ni2P) was synthesized by H2-TPR method at a high temperature of 650 ℃; Secondly, the resulting undoped Ni2P/SiO2catalyst were doped by Co, Fe and Mo by in-situ phosphitylation method at a low temperature of 300 ℃, respectively.
Characterization results indicate that, the introduction of the three doping metals results in a homogeneous Ni2P-CoP, Ni2P-FeP, Ni2P-MoP mixtures, rather than bimetallic alloying phases, and meanwhile the electronic structure of Ni2P was well tuned by the three doping agents, thus laying a foundation to accurately study the electronic effects of Ni2P catalysts on the HDS functions.
Further characterization results show that, the three doping metals greatly influence the electron density of Ni2P sites, and their electron-deficiency followed the following order: Mo-Ni2P/SiO2>Ni2P/SiO2>Co-Ni2P/SiO2>Fe-Ni2P/SiO2. 4,6-DMDBT HDS performance assessment results indicate a volcano-type relationship between HDS intrinsic activity and electronic structures of Ni2P sites. A medium value of electron density of Ni2P optimizes the appropriate interaction between sulfur organic compounds and active sites, thereby Co-Ni2P/SiO2with moderate electron density value has the best HDS activity. Furthermore, the electron-deficiency of Ni2P sites significantly enhances the DDS selectivity. Compared with Ni2P/SiO2catalyst, Fe-Ni2P/SiO2and Co-Ni2P/SiO2with higher electron density in Ni2P sites hold an enhanced HYD selectivity, while Mo-Ni2P/SiO2catalyst with higher electron-deficiency of Ni2P exhibits an enhanced DDS selectivity. It is rational and effective to modify the electronic structure and density of states of Ni2P sites to enhance the promoting effect and thereby improve the catalytic performance.
Acknowledgement
The authors thank Dr. Hui Yuan for their help in sample analysis and assessment.
猜你喜欢
无机化学学报(2022年10期)2022-10-10
飞言情A(2021年7期)2021-09-26
今日农业(2020年13期)2020-12-15
无机化学学报(2020年1期)2020-02-11
辽金历史与考古(2019年0期)2020-01-06
校园英语·中旬(2019年11期)2019-11-26
无机化学学报(2019年8期)2019-08-08
少先队活动(2018年7期)2018-12-29
走向世界(2018年11期)2018-12-26
走向世界(2018年11期)2018-12-26
