
B族链球菌基因cpsG抑制人类宫颈癌细胞增殖的研究
2022-07-19 01:04李艳娜王莘童苏顺海
同济大学学报(医学版) 2022年3期
李艳娜, 支 恒, 王莘童, 苏顺海, 徐 磊
(同济大学医学院,上海 200092)
宫颈癌是世界上女性第二大常见恶性肿瘤,也是导致女性癌症死亡的主要原因之一[1]。宫颈癌及其相关调控机制的研究尚待进一步深入。近年来,靶向治疗宫颈癌显示出前所未有的优势。为了改善宫颈癌患者的预后,仍然迫切需要开发更有前景的治疗靶点[2]。B族链球菌(Group B streptococcus, GBS)是严重的新生儿感染的主要原因。GBS是一种人体内的机会致病菌,是肠道和阴道正常菌群的一部分,母体定植是GBS传播的主要途径。目前,已确认的血清型多达10种(Ⅰa、Ⅰb和Ⅱ~Ⅸ)[3]。GBS经常寄生于孕妇,并可在婴幼儿中引起败血症和脑膜炎。如果通过母亲免疫来预防定植,可能会减少围产期GBS引起的相关疾病。B族链球菌荚膜多糖(capsular polysaccharide, CPS)是一种重要的毒力因子,也用于GBS分型。所有合成CPS所需的基因都在B族链球菌CPS操纵子上,其中包含一个高度可变的cps决定区域(cpsG-cpsK)。研究表明,GBS能释放毒素导致宫颈炎症发生等,但是GBS中每个基因如何对宫颈细胞发挥确切作用尚不清楚。GBS感染细胞后,GBS的基因组暴露在细胞中,并利用细胞内的转录翻译机器触发GBS相关基因的表达并发挥独立的作用[4]。然而目前尚未有B族链球菌相关基因cpsG与人类癌症发生相关的报道。本研究将GBS的关键基因cpsG的表达序列构建到慢病毒中,将慢病毒感染人类宫颈癌细胞,使得cpsG在宫颈癌细胞中表达,通过体外生长以及高通量RNA测序探索cpsG影响人类宫颈癌细胞某些转录组功能的可能机制。
1 材料与方法
1.1 实验用细胞
人类宫颈癌细胞(HPV18+)购自中科院上海细胞所。
1.2 实验试剂
细胞培养用胎牛血清(FBS)、DMEM液体培养基、PBS、胰酶购自上海欲立生物科技有限公司;rLV-cpsG-HA-ZsGreen慢病毒和rLV-ZsGreen对照慢病毒购自武汉维诺赛生物技术有限公司;嘌呤霉素、增强型CCK-8试剂盒;抗HA抗体(51064-2-AP)购自武汉三鹰生物技术有限公司;RNA测序由北京诺禾致源科技股份有限公司完成;引物由苏州金唯智生物科技有限公司合成;PAGE凝胶快速制备试剂盒购自上海雅酶生物医药科技有限公司;通用型ECL发光底物试剂盒购自上海圣尔生物科技有限公司。
1.3 方法
1.3.1 人类宫颈癌细胞培养 将HPV18+培养于含10%FBS的DMEM完全培养基中,且在饱和湿度以及含5%CO2和37 ℃的细胞培养箱中进行培养。每隔3 d进行1次换液。
1.3.2 慢病毒感染和稳定细胞系构建 胰酶消化处于对数生长期的细胞,按照要求进行慢病毒感染,48 h后更换培养液。1周后采用嘌呤霉素(3 mg/L)持续筛选,最后用进行qRT-PCR和Western印迹法检测感染效率。
1.3.3 提取细胞总RNA进行RT-PCR 提取细胞总RNA,反转录为cDNA,进行PCR反应,以β-actin为内参基因,反应条件: 94 ℃ 3 min,94 ℃ 30 s,55 ℃ 30 s,72 ℃ 30 s,72 ℃ 10 min,39个循环。引物序列如下。cpsG P1: 5′-GGACACATGAACAGCAGTTC-3′;cpsG P2: 5′-TTTCAACGCTTCCGCAAGTC-3′。
1.3.4 Western印迹法检测 取离心后的细胞,加入RIPA裂解液冰上放置30 min,待细胞完全裂解后,12 000 r/min,离心半径650 mm,4 ℃离心20 min。将提取的蛋白质溶液加入蛋白上样缓冲液100 ℃加热10 min进行变性。12%SDS-PAGE电泳1.5 h,使用PVDF膜转膜20 min,用含10%脱脂奶粉的TBST 37 ℃封闭2 h,一抗HA 1∶1 500,β-actin 1∶2 000,4 ℃ 孵育过夜,TBST洗涤3次,每次5 min,二抗1∶3 000,37 ℃ 2 h,TBST洗涤3次,每次5 min,用ECL进行发光反应进行拍照。
1.3.5 CCK8检测 分别取对照组和实验组细胞,接种于96孔板中,每孔接种3×103个细胞,设置4行平行复孔,分别于第0、1、2、3天每孔加入CCK8溶液10 μL;5%CO2,37 ℃孵育4 h后,在多功能酶标仪检测450 nm波长处的光密度(D450)数值。
1.3.6 集落形成实验 分别取对照组和实验组细胞,接种于6孔板中,每孔300个细胞,设置3个复孔,培养2周后,弃培养液,用PBS冲洗1次,弃上清液,最后每孔加用0.5%结晶紫染色,在摇床上放置,50~60 r/min,2~3 h后弃上清液,水洗,晾干后拍照计数。
1.3.7 RNA测序 提取细胞总RNA,通过电泳鉴定RNA的完整性,由北京诺禾致源科技股份有限公司进行RNA测序,并进行生物信息学分析。
1.4 统计学处理
2 结 果
2.1 过表达cpsG基因的稳定细胞系的筛选
为了研究cpsG对人类宫颈癌细胞的影响,通过人工合成的方法将cpsG的读码框序列(ORF)融合HA标签连接到质粒pLVX-mCMV-ZsGreen的限制性内切酶EcoRⅠ和BamHⅠ之间。将质粒进行酶切和琼脂糖电泳鉴定,证明质粒中插入了cpsG-HA。最后通过测序证明质粒中插入了正确的cpsG-HA,提示成功构建了pLVX-cpsG-HA-mCMV-ZsGreen质粒。将pLVX-cpsG-HA-mCMV-ZsGreen质粒转染HEK293细胞,24 h后HEK293细胞能成功表达绿色荧光蛋白。进一步利用HEK293细胞制备慢病毒rLV-cpsG-HA-ZsGreen: 将1 μL rLV-cpsG-HA-ZsGreen慢病毒感染HEK293细胞72 h 后能表达绿色荧光蛋白。提示成功制备了过表达cpsG的慢病毒。将慢病毒rLV-cpsG-HA-ZsGreen感染人类宫颈癌细胞,用嘌呤霉素筛选,并在荧光显微镜下挑取表达GFP的单克隆细胞,并扩大培养(图1A)。然后提取细胞的总RNA,RT-PCR结果显示: 与rLV对照组相比,rLV-cpsG组产生了过量的cpsG-HA转录产物(图1B)。提取2组细胞的总蛋白,进行Western印迹法检测,结果显示与rLV对照组相比,rLV-cpsG组产生了过量的cpsG-HA融合蛋白(图1C)。上述结果提示,成功构建了过表达cpsG的稳定细胞系。
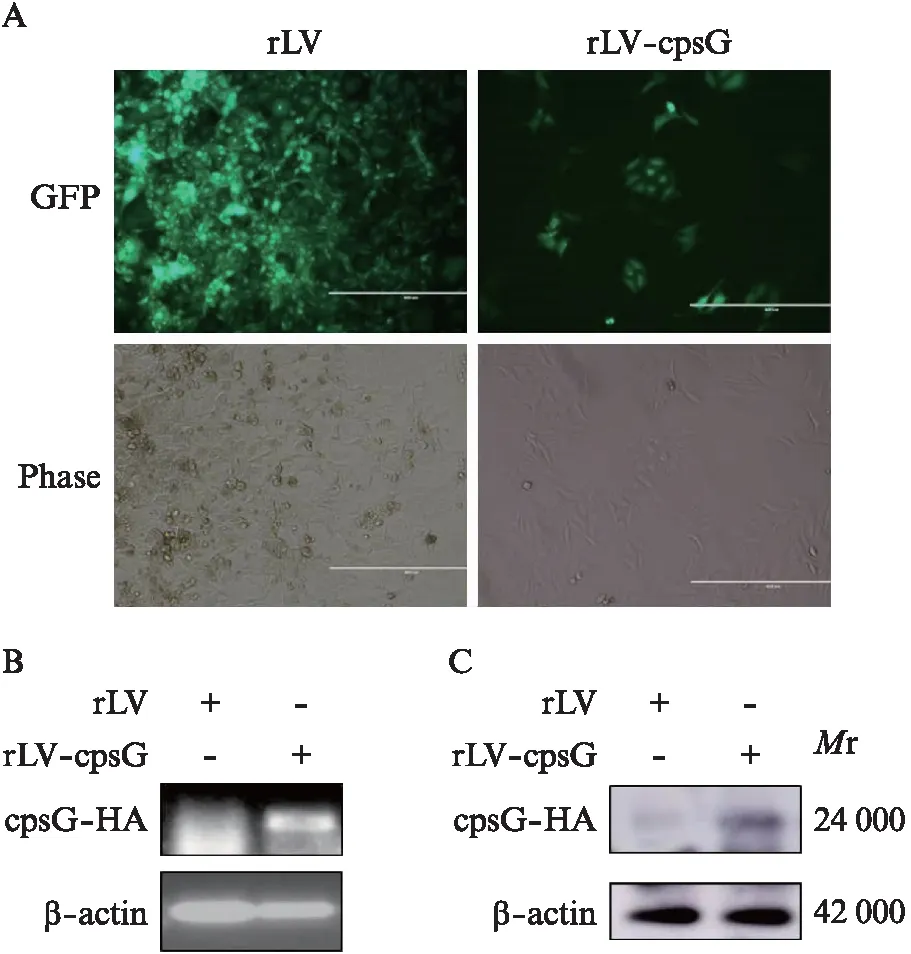
图1 过表达cpsG的稳定细胞系筛选Fig.1 Screening of stable cell lines overexpressing cpsGA: rLV-cpsG-HA-ZsGreen慢病毒感染人类宫颈癌细胞(×100);B: RT-PCR扩增结果;C: Western印迹法结果
2.2 cpsG基因抑制人类宫颈癌细胞体外生长
为了探讨cpsG基因对人类宫颈癌细胞体外生长的影响,对上述的稳定细胞系进行生长曲线和集落形成能力分析。首先,利用CCK8的方法对细胞增殖能力进行测定,2组相比差异有统计学意义(P<0.01),显示cpsG能够抑制细胞的体外增殖能力,见图2。细胞平板集落形成能力测定,将每组的300个细胞分别接种到六孔板,每组3个复孔,10 d后用结晶紫染色,并进行计数分析。结果显示: rLV组的平板集落形成率为(41.1±3.57)%,rLV-cpsG组的平板集落形成率为(15.77±2.157)%,2组相比差异有统计学意义[(41.1±3.57)%vs(15.77±2.157)%,P=0.001 2],显示cpsG抑制了细胞的集落形成能力。上述结果提示cpsG基因抑制人类宫颈癌细胞体外生长。

图2 cpsG基因抑制人类宫颈癌细胞体外生长分析Fig.2 Inhibition of human cervical cancer cell growth by cpsG gene in vitro*P<0.05,**P<0.01
2.3 cpsG改变了人类宫颈癌细胞的基因转录能力
为了研究cpsG对人类宫颈癌细胞基因转录能力的影响,本研究提取2组细胞的总RNA进行电泳分析和Agilent5400峰图分析,显示完整的28 S、18 S、5 S。然后进行了高通量RNA测序分析。对两个样本分别进行基因表达水平的定量分析,包括基因表达分布,计算各样本所有基因的表达值(FPKM)后,通过盒形图展示不同样本基因表达水平的分布情况,见图3A。样本间的相关性分析显示皮尔逊相关系数的平方(R2)为0.854(R2>0.8),见图3B。基因的密度分布分析显示2组的基因分布密度是有差异的,见图3C、3D。在rLV组和rLV-cpsG组中,差异基因韦恩图显示1 276个基因仅出现在rLV组,931个基因仅出现在rLV-cpsG组,11 596个基因在2组中都出现(图4A)。差异基因数目统计分析(图4B)、火山图分析(图4C)显示2组中上调基因有490个,下调基因有1 195个,22 257个基因无显著变化。上述结果提示cpsG改变了人类宫颈癌细胞中部分基因的转录水平。
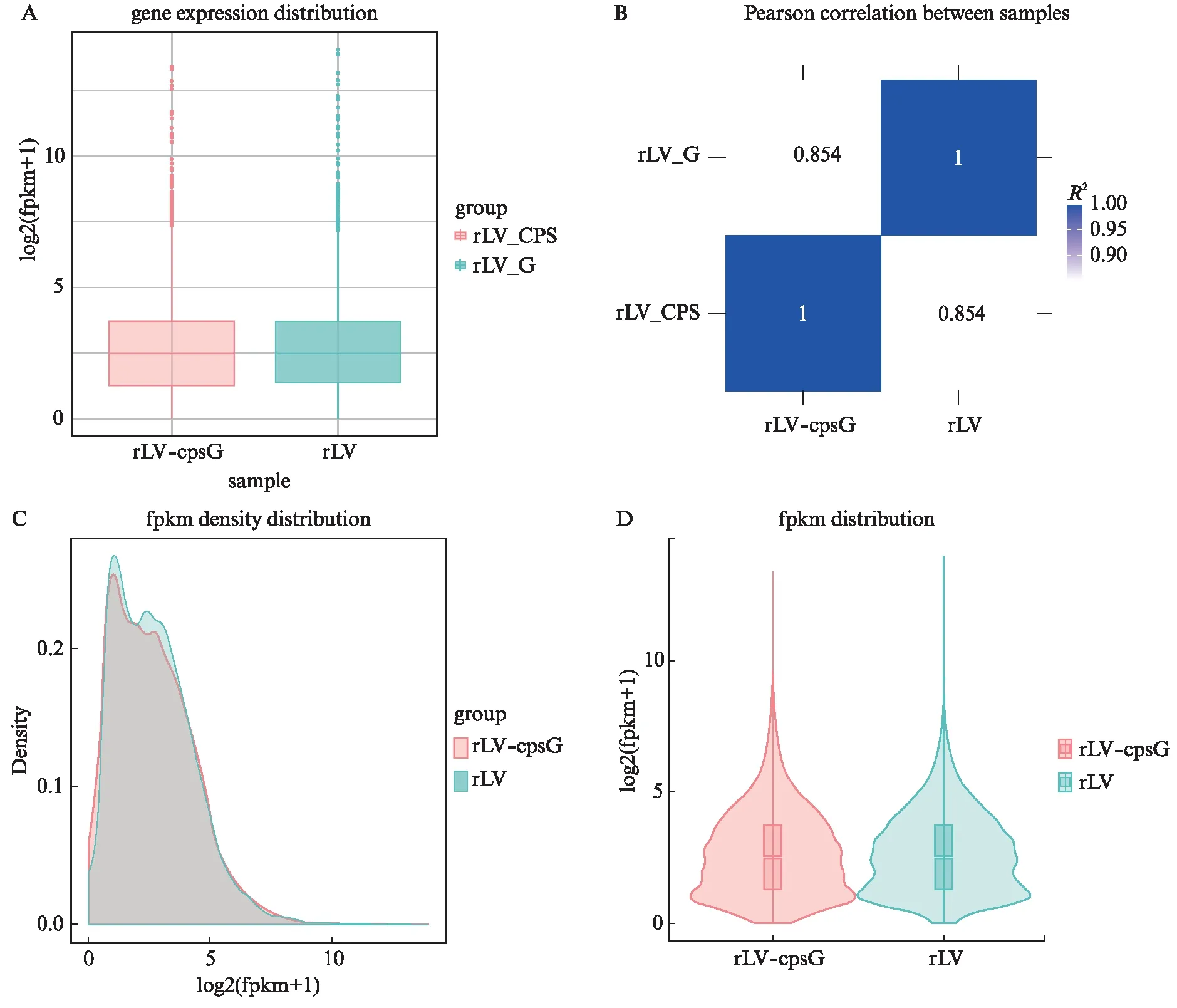
图3 基因表达水平的定量分析Fig.3 Quantitative analysis of gene expression levelA: 样本基因表达量分布盒形图;B: 样本间相关性热图;C: FPKM密度分布;D: FPKM分布violin图
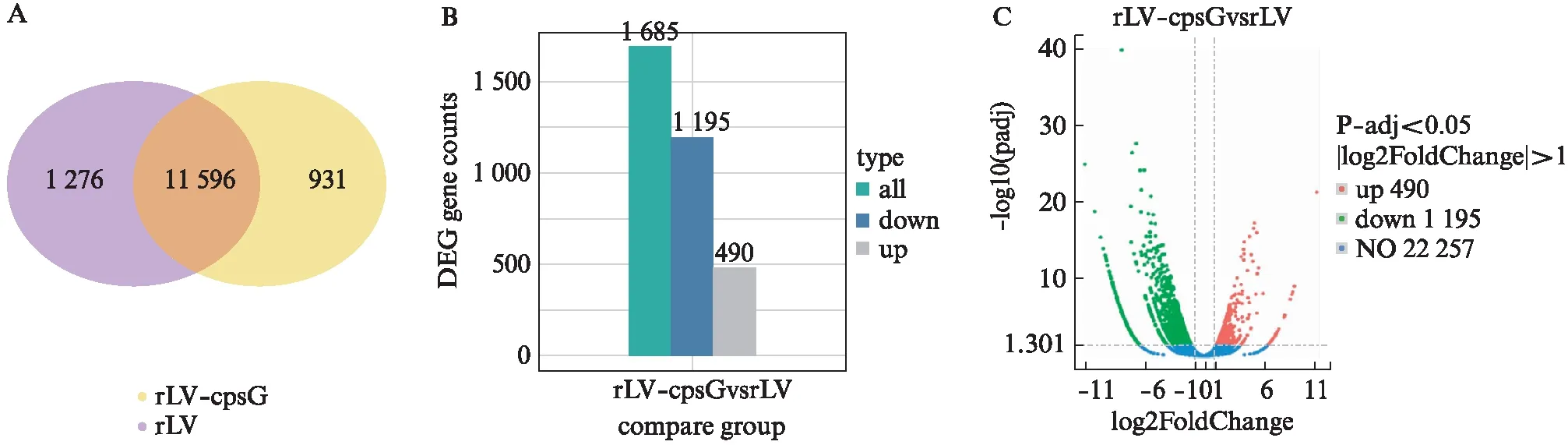
图4 基因差异分析Fig.4 Analysis of gene differencesA: 差异基因韦恩图,韦恩图中圈内所有数字之和代表该比较组合差异基因总数,重叠区域表示组合间共有的差异基因个数,不同的颜色表示不同的比较组合;B: 差异基因数目统计柱状图;蓝色和灰色分别表示上调和下调的差异基因,柱子上的数字表示差异基因数目;C: 差异基因的火山图
2.4 cpsG改变了人类宫颈癌细胞的基因功能
既然cpsG改变了人类宫颈癌细胞部分基因转录能力,将进一步分析cpsG对人类宫颈癌细胞中相关基因功能的影响,包括基因本体论(GO)分析、京都基因和基因组百科全书(KEGG)分析。GO富集分析结果显示: 与rLV对照组相比,rLV-cpsG组中重要的下调GO、BP、CC和MF;重要的上调的GO、BP、CC和MF见图5。提示cpsG基因影响了人类宫颈癌细胞中基因的GO功能。
KEGG分析显示: 与rLV对照组相比,rLV-cpsG组中有显著意义的下调的KEGG通路见图5A;有显著意义的重要的上调的KEGG通路见图6。结果提示cpsG影响了人类宫颈癌细胞中一些基因的KEGG通路。
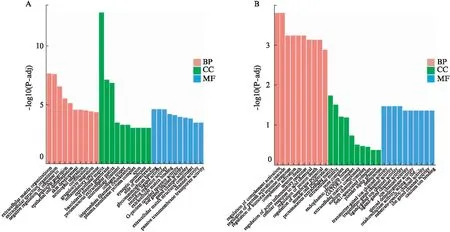
图5 基因功能富集分析Fig.5 Enrichment analysis of gene functionA: GO富集分析散点图,横坐标为注释到GO Term上的差异基因数与差异基因总数的比值,纵坐标为GO Term,点的大小代表注释到GO Term上的基因数,颜色从红到紫代表富集的显著性大小,a: 下调,b: 上调。B: GO富集分析柱状图。横坐标为GO Term,纵坐标为GO Term富集的显著性平,用-log10(padj)表示,BP: 生物过程;CC: 细胞组分;MF: 分子功能;a: 下调,b: 上调

图6 KEGG富集分析柱状图Fig.6 KEGG analysis横坐标为KEGG通路,纵坐标为通路富集的显著性水平;A: 下调,B: 上调
2.5 cpsG调控了人类宫颈癌细胞基因的融合
融合基因分析显示,rLV组中融合基因包括MT-ATP6-MT-ATP8、B3GAT3-MT-ATP8、KANSLI-ARL17B、GALNTI-CCBE1、GNB1-NADK,见图7A。rLV-cpsG组中融合基因包括MT-ATP6-MT-ATP8、GALNT1-CCBE1、GNB1-NADK、B3GAT3-GANAB、LEPROT-LEPR,见图7B。其中MT-ATP6-MT-ATP8、GALNT1-CCBE1、GNB1-NADK、SREBF2-MEI1、HN-RNPUL2-C11orf49既出现在rLV对照组中,又出现在rLV-cpsG组中;结果提示cpsG改变了人类宫颈癌细胞的部分基因的功能。

图7 融合基因circos图分析Fig.7 Circos plot analysis of fusion geneA: 对照组;B: rLV-cpsG组
3 讨论
本研究发现B族链球菌相关基因cpsG能够抑制人类宫颈癌细胞的增殖能力,揭示cpsG可能与细胞中部分基因的转录组学改变有关。
3.1 cpsG基因与人类宫颈癌发生呈负相关
本研究结果提示cpsG基因能够显著抑制人类宫颈癌细胞体外生长能力,细胞生长曲线的测定和细胞集落形成能力测定证实这一结论。cps是一种重要的毒力因子,也可用于GBS分型[4]。尽管cpsG主要与一些侵袭性疾病有关[3],然而目前尚未有关于B族链球菌相关基因cpsG与人类癌症相关的报道,本研究证实了cpsG能够抑制宫颈癌细胞增殖能力,推测cpsG可能通过改变一些肿瘤相关基因的功能和信号通路,从而抑制人类宫颈癌细胞的进展。
3.2 cpsG改变了人类宫颈癌细胞中基因的转录能力
本研究通过高通量RNA测序技术,发现cpsG能够调控人类宫颈癌细胞部分基因的转录能力。研究表明,上述cpsG上调的基因中DSC3、STXBP5L、IGFBP4、FAM107A与肿瘤的发生有关[5-8]。DSC3在乳腺癌、前列腺癌和结直肠癌细胞中下调表达,特别是DSC3基因的DNA甲基化状态是结直肠癌的预后标志之一[5],提示在一些肿瘤中DSC3可能是一个抑癌基因,推测cpsG能够增强DSC3的表达而发挥抑癌作用。研究发现STXBP5L能形成环状的circ-STXBP5L,并且其在小细胞肺癌中上调表达和促进小细胞肺癌的发生[6]。本研究中cpsG能促进STXBP5L的转录能力,提示STXBP5L可能在cpsG的作用下在宫颈癌细胞中发挥相反的作用。研究表明IGFBP4缺失会加速肺癌和肝癌的发生[7],提示IGFBP4是一个抑癌基因,本研究中cpsG能促进IGFBP4的转录能力,推测cpsG能够增强IGFBP4的表达而发挥抑癌作用。研究发现FAM107A在非小细胞肺癌患者中的表达降低,FAM107A在喉癌细胞系和原发肿瘤中完全下调[8]。本研究中cpsG能够促进FAM107A转录能力,提示FAM107A在cpsG的作用下抑制宫颈癌细胞生长。上述推测均有待实验进一步证实。
本研究中发现的在人类宫颈癌细胞中能被cpsG下调的基因KRT6A、KRT17、WFDC2与肿瘤的发生有关[9-11]。研究显示KRT6A促进肺癌的增殖和转移[9]。本研究中cpsG能抑制KRT6A在转录水平的表达,提示cpsG可能通过抑制KRT6A的转录而发挥抑癌作用。研究发现KRT17在宫颈癌细胞中过表达[10]。而cpsG能抑制KRT17的转录能力,提示cpsG可能通过抑制KRT17的转录而发挥抑制宫颈癌细胞生长的作用。研究发现WFDC2在卵巢癌中促进增殖和转移[11]。本研究中cpsG能抑制WFDC2的转录能力,提示cpsG可能通过抑制WFDC2的转录而发挥抑癌作用。上述推测均有待实验进一步证实。
3.3 cpsG调节了人类宫颈癌细胞中的一些基因的功能和信号通路
本研究中通过基因本体论(GO)分析和京都基因和基因组百科全书(KEGG)分析清楚地发现cpsG调节了人类宫颈癌细胞中的一些基因的功能和信号通路。(1) 被cpsG下调的GO,主要包括G蛋白偶联受体活性、上皮细胞分化、受体配体活性。(2) 被cpsG上调的GO,主要包括蛋白活化级联调控、Seh1相关复合物。(3) 被cpsG下调的KEGG通路,主要包括PI3K-Akt信号通路、TNF信号通路、NF-kappa B信号通路、RAS信号通路。(4) 被cpsG上调的KEGG通路,主要包括脂肪酸降解通路、p53信号通路。研究发现PI3K-Akt信号通路有着广泛的促癌作用,其在前列腺癌、胃肠道肿瘤、乳腺癌和宫颈癌中调节肿瘤细胞转移和侵袭,宫颈癌组织中PI3k基因及蛋白水平随肿瘤恶性程度的增加而升高[12-13]。本研究中发现cpsG能显著抑制PI3K-Akt信号通路,提示cpsG可能通过阻碍该通路而起到抑制宫颈癌的发生。研究表明TNF是一种主要的炎性细胞因子,具有诱导实验性癌症快速出血性坏死的能力,TNF-α是乳腺癌患者肿瘤微环境中发现的重要促炎细胞因子之一,在乳腺癌进展和转移过程中的致瘤作用[14]。而cpsG抑制了TNF通路的活性,提示cpsG可能通过抑制其活性起到抑癌的作用。研究发现NF-κB信号通路与许多类型的肿瘤有关,NF-κB是许多基因的转录因子,也成为各种人类癌症的主要罪魁祸首,主要是因为它有保护转化细胞逃逸凋亡的能力[15]。本研究中cpsG抑制了该通路的活性,提示cpsG可能通过抑制其活性起到抑癌作用。研究发现P53信号通路在肿瘤发生发展中起抑制作用,p53发生突变导致细胞异常增殖和肿瘤进展,并加剧癌细胞的恶性特性[16]。而cpsG上调该通路,提示cpsG可能通过影响该通路活性起到抑癌作用。cpsG可能通过影响了人类宫颈癌细胞中一些基因的功能和信号通路而抑制宫颈癌细胞的生长。待进一步实验证明。
3.4 cpsG通过调控基因的融合抑制人类宫颈癌细胞的生长
本研究中融合基因分析提示: (1) 只出现在rLV组中融合基因,包括B3GAT3-ATP8、SLC39A10-SESTD1、TMEM132A-TMEM109。(2) 只出现在rLV-cpsG组中融合基因,包括B3GAT3-GANA、MRPS6-SON、SLC5A3-SON。研究表明融合基因可以导致原来基因功能的丧失,或者增强了原来基因的功能,或者产生了新的基因功能。B3GAT3高表达与肝癌预后不良有关,ATP8突变与乳腺癌发生相关[17],B3GAT3和ATP8形成融合基因后其功能可能被抑制或加强。而cpsG能够抑制B3GAT3-ATP8融合基因的形成,推测cpsG可能导致B3GAT3-ATP8功能的丧失而抑制宫颈癌细胞的生长。研究发现MRPS6蛋白在乳腺癌中过表达,SON在胰腺癌中过表达[18]。而cpsG能够促进MRPS6-SON融合基因的形成,推测cpsG可能导致MRPS6-SON原来基因功能的丧失,产生新的功能而抑制宫颈癌细胞的生长。研究提示cpsG通过调控基因的融合抑制人类宫颈癌细胞的生长。
总之,本研究提示cpsG抑制了人类宫颈癌细胞生长,主要通过上调DSC3、IGFBP4、PLCL1和下调KRT6A、KRT17、WFDC2基因的转录,从而抑制了G蛋白偶联受体活性、PI3K-Akt信号通路、TNF信号通路、NF-κB信号通路和激活了脂肪酸降解、p53信号通路。有趣的是,cpsG能影响B3GAT3-ATP8、LEPROT-LEPR、MRPS6-SON融合基因的形成。本研究为人类宫颈癌的临床诊断和治疗提供了理论依据。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国医学物理学杂志(2022年9期)2022-10-09
中国现代医生(2022年19期)2022-08-25
海峡姐妹(2020年2期)2020-03-03
保健与生活(2019年3期)2019-08-01
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
江苏农业科学(2017年5期)2017-04-15
中国民族民间医药·下半月(2014年4期)2014-09-26
中国民族民间医药·下半月(2014年4期)2014-09-26
