
Wong型无肌病性皮肌炎一例
2022-07-13 13:59AlizaPaudyal吕小岩
中国麻风皮肤病杂志 2022年9期
郑 铭 Aliza Paudyal 吕小岩
四川大学华西医院皮肤性病科,四川成都,610041
临床资料患者,女,72岁。因头面颈部红斑、丘疹伴瘙痒16年就诊。患者16年前无明显诱因出现头面颈部红斑、丘疹,伴瘙痒,日晒后加重,无四肢乏力、吞咽困难、呼吸困难等。曾于外院行皮肤活检术,诊断为“皮肌炎”,具体治疗不详,皮疹未见好转。既往有糖尿病、高血压病史,个人史和家族史无特殊。
体检:四肢肌力Ⅴ级,各系统未查见明显异常。皮肤科检查:双上眼睑水肿性紫红色斑(图1a),额部见黄红色毛囊性角化过度性丘疹和融合性斑块,部分丘疹顶端可见角栓(图1b),颈胸部V型区(图1c)、项部和肩背部紫红斑(图1d)。
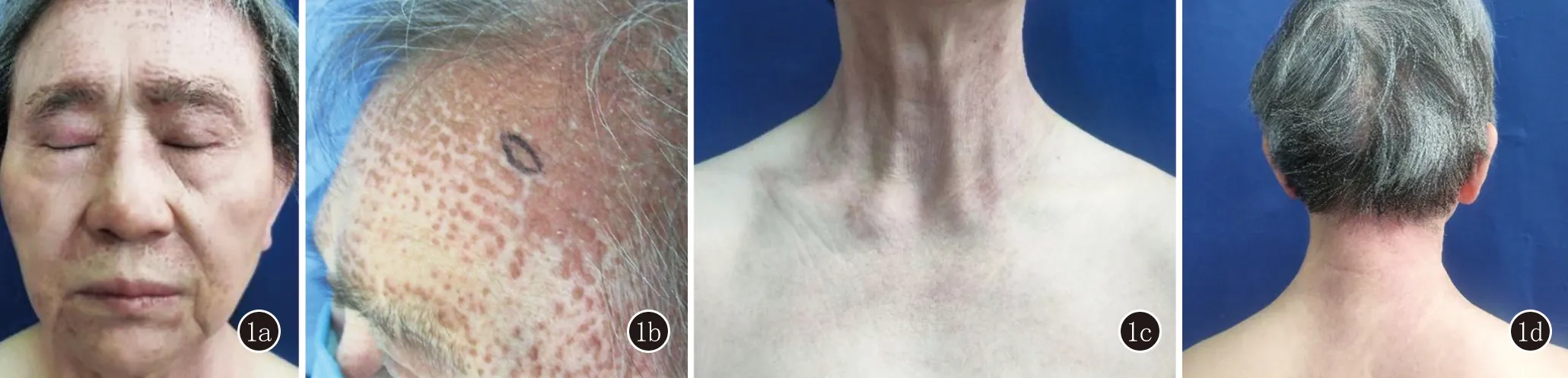
图1 1a: 双上眼睑水肿性紫红色斑;1b: 额部黄红色毛囊性角化过度性丘疹和融合性斑块,部分丘疹顶端可见角栓;1c: 颈部、胸前区紫红斑;1d:项部、肩背部紫红斑
辅助检查:抗核抗体1∶100 颗粒型;肌炎特异性抗体:抗TIF1-γ抗体IgG阳性;血常规、肝肾功、肌酶、肿瘤标志物等未见异常。胸部CT、腹部彩超、泌尿系彩超、妇科彩超未见异常。额部皮肤组织病理:表皮明显角化过度(图2a),局灶性角化不全(图2b),部分基底层液化变性(图2c),真皮浅层小血管周围及毛囊皮脂腺周围中等量淋巴细胞浸润。

图2 2a:表皮明显角化过度,真皮浅层小血管周围中等量淋巴细胞浸润(HE,×100);2b:局灶性角化不全(HE,×400);2c:基底层液化变性(HE,×400)
诊断:Wong型无肌病性皮肌炎。治疗:阿维A胶囊20mg/d、海棠合剂口服,外用1%吡美莫司乳膏。治疗1月后,患者额部丘疹、斑块变平,颜色变浅,未见新发皮损。目前仍在随访中。
讨论Wong型皮肌炎是由Wong在1969年提出的一种罕见的皮肌炎亚型,其皮肤表现除经典皮肌炎的皮损外,还可出现毛囊角化过度性丘疹、融合性斑块、掌跖角皮症等毛发红糠疹样皮损,任何部位均可发生[1,2]。Wong型皮肌炎的皮肤组织病理表现多样,可表现为皮肌炎的病理改变,如界面皮炎、真皮层黏液沉积,血管和附属器周围炎症细胞浸润等,也可表现为水平方向和垂直方向交替出现的局灶性角化过度和角化不全等符合毛发红糠疹的病理改变,亦有部分Wong型皮肌炎的皮肤病理改变界于两者之间[2,3]。Wong型皮肌炎的肌肉表现和经典皮肌炎无明显差异,轻者仅有发音障碍,重者可有呼吸肌受累,还有患者表现为无肌病性皮肌炎[3-5]。Wong型皮肌炎亦可伴发肺间质病变或恶性肿瘤[1,2,5-7]。治疗可选择羟氯喹、糖皮质激素、硫唑嘌呤等,最近有联合使用利妥昔单抗和人免疫球蛋白成功治疗Wong型皮肌炎的个案报道[8]。
本例患者有Heliotrope征、V字征、披肩征等符合皮肌炎皮肤表现的皮疹,同时伴有瘙痒,组织病理学改变符合皮肌炎,因此符合Sontheimer无肌病性皮肌炎诊断标准[9]。因为患者同时伴有毛发红糠疹样皮疹,故诊断为Wong型无肌病性皮肌炎。本病需要与毛囊角化病、毛发红糠疹等疾病鉴别。毛囊角化病为常染色体显性遗传性皮肤病,发病年龄多在6~20岁,临床表现为红色至褐色的角化性丘疹,分布倾向于皮脂溢出部位,部分皮损表面附有痂皮,常伴有恶臭,组织学特征为棘层松解和角化不良[10],本例为老年女性,病程已有16年,且合并皮肌炎的皮肤表现。典型的毛发红糠疹有自限性,多在3~5年内痊愈[10],因此不考虑毛发红糠疹。
临床中如果见到不典型的毛发红糠疹患者,可以寻找其他的皮疹线索,考虑到特殊类型皮肌炎的可能。因为患者抗TIF1-γ抗体阳性,该抗体被认为是成人皮肌炎患者合并恶性肿瘤的独立危险因素[11],所以目前患者仍在密切随访中。
猜你喜欢
纺织高校基础科学学报(2022年2期)2022-07-29
医学前沿(2021年6期)2021-09-10
数学学习与研究(2021年18期)2021-08-06
家庭科学·新健康(2020年6期)2020-07-06
中国美容医学(2019年2期)2019-03-13
家庭医学·下半月(2018年8期)2018-10-17
东方教育(2017年14期)2017-09-25
飞碟探索(2017年8期)2017-08-11
课程教育研究·学法教法研究(2016年1期)2016-03-17
故事作文·低年级(2009年4期)2009-04-21
