
显性营养不良型大疱性表皮松解症COL7A1基因突变分析
2022-07-13 13:59谢锦莹周顺婷黄闽嘉罗志强周书文
中国麻风皮肤病杂志 2022年9期
祝 玉 吴 玮 谢锦莹 周顺婷 黄闽嘉 罗志强 周书文
广东医科大学附属医院皮肤科,广东湛江,524000
营养不良型大疱性表皮松解症(epidermolysis bullosa,EB)是一组以皮肤和(或)黏膜脆性增加为特征的罕见遗传病,可发生于局部或全身。它是由编码不同蛋白质的基因突变引起的,这些蛋白质主要与真皮-表皮连接的结构和功能有关。根据皮肤裂隙水平(从上到下)分为四种类型:单纯型(EBS,epidermolysis bullosa simplex)、交界型(JEB,junctional epidermolysis bullosa)、营养不良型(DEB)和Kinder综合征(KS)。分子遗传学表明,位于染色体3p21的COL7A1基因(C7)为DEB的致病基因[1]。本文通过基因测序确诊2例来自一个广东DEB家系的患者,并发现了位于86号外显子的基因突变位点。
1 临床资料及家系调查
先证者,女,广东廉江人,51岁。双胫前反复结节、水疱,伴瘙痒40余年就诊。患者自10岁以来双胫前反复出现丘疹和水疱,慢慢演变成结节和色素沉着,瘙痒明显,逐渐双上肢也出现类似皮损。皮肤科情况:双侧小腿见密集分布孤立暗红色结节,部分中央结痂,见少部分丘疹,无水疱。双侧大腿可见散在红色丘疹、结节,部分中央结痂,无水疱。双上肢可见散在红色丘疹,部分表面结痂。趾甲可见甲增厚、表面凹凸不平和甲萎缩(图1)。组织病理:正角化过度,表皮略肥厚,表皮突消失,见表皮下裂隙,真皮浅层胶原纤维增生致密,见少许淋巴组织细胞和浆细胞浸润(图2)。免疫荧光:基底膜带IgG、IgA、IgM及C3均阴性。初步诊断为大疱性表皮松解症。予阿维A胶囊20 mg日2次,复方甘草酸苷片50 mg日3次,复方醋酸曲安奈德溶液及卤米松/三氯生乳膏外用治疗,效果不佳,目前正在随访中。

图1 先证者临床照片 1a、1b:双上肢散在多发丘疹,部分中央结痂,伴脱屑;1c、1d:双侧小腿可见密集分布红色丘疹、结节,部分融合。双侧大腿可见散在多发红色丘疹,部分中央结痂;1e:趾甲可见甲增厚、表面凹凸不平和甲萎缩 图2 先证者组织病理:正角化过度,表皮突消失,见表皮下裂隙,真皮浅层胶原纤维增生致密(HE,×200)
先证者胞妹,44岁,双下肢丘疹、结节30余年,伴轻微瘙痒。患者自10岁以来双下肢丘疹、结节,伴轻微瘙痒。皮肤科情况:双侧小腿和足背可见密集分布类圆形丘疹、结节,部分呈线状排列,背部和右侧肘关节伸侧见散在多发皮色扁平丘疹,无水疱。10趾甲可见甲增厚、表面凹凸不平和甲萎缩(图3)。
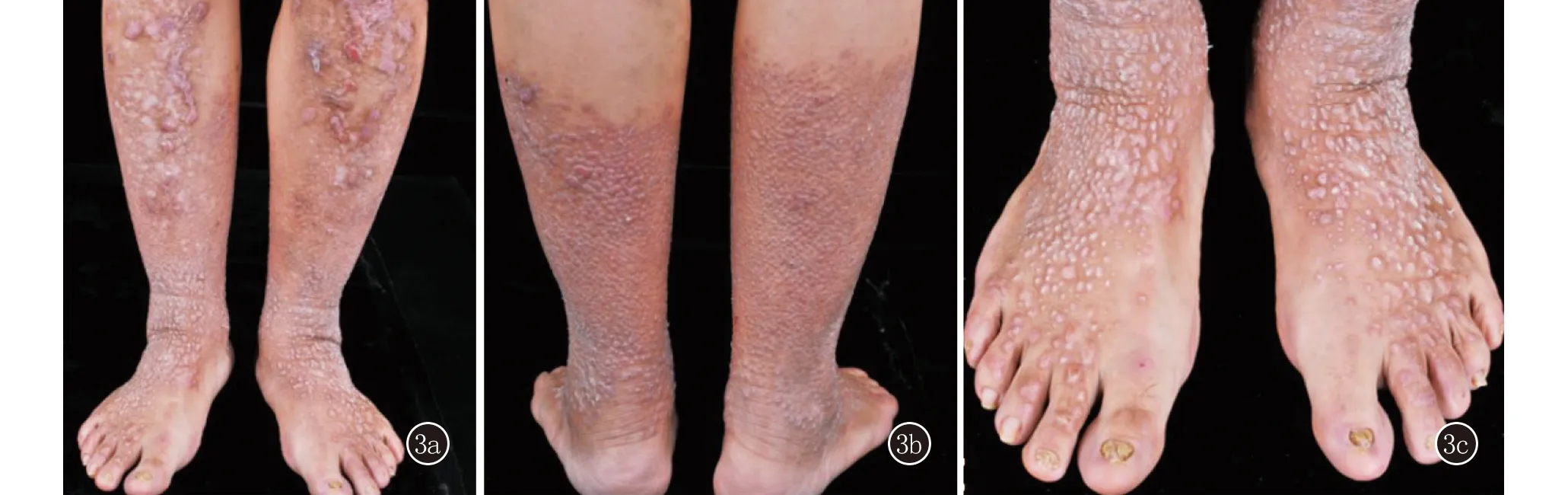
3a、3b:双侧小腿密集分布类圆形丘疹、结节,部分融合成线状,双侧胫前可见片状色素减退;3c:双足背可见密集分布类圆形丘疹、结节,双侧趾甲可见甲营养不良
先证者的父亲和胞妹的儿子亦有类似症状,母亲、其余两个姐妹和其子女无类似症状。该家系4代20名成员,发现有4例患者(图4),父母否认近亲结婚。根据家系调查结果,该病的遗传方式为常染色体显性遗传。

4a:家系图(4代20名成员,发现有4例患者,分别为先证者及其父亲、妹妹和外甥),箭头示先证者;4b:先证者及其胞妹测序分析提示:杂合突变,位于外显子86上的c.6761G>A;p.Gly2254Glu(上方为先证者,下方为其胞妹)
2 基因检测
2.1 经患者知情同意后,取患者及其胞妹的外周静脉血各6 mL,标本送至深圳华大临床检验中心,采用芯片捕获高通量方法对先证者进行DNA基因全外显子直接测序。检测区域:人类基因组中约2万个基因的外显子区;检测策略:针对受检者主诉,对OMIM(0nline Mendelian Inheritance in Man)数据库收录的明确致病关系基因进行分析;检测方法:芯片捕获高通量测序。
2.2 对先证者及其胞妹的外周血DNA用Sanger测序验证突变位点。
3 结果
3.1 先证者 在先证者COL7A1基因外显子86上检出一个与受检者临床表型相关的意义未明变异c.6761G>A;p.Gly2254Glu(图4),根据ACMG(American College of Medical Genetics and Genomics)指南,该变异被判断为意义未明变异。采用Sanger测序验证突变位点的结果显示为一杂合突变。
3.2 患病胞妹 采用Sanger测序验证突变位点的结果与先证者一致:c.6761G>A;p.Gly2254Glu,为杂合突变。
4 讨论
EB是指皮肤或黏膜受到轻微外伤后即可引起水疱的一组异质的罕见遗传性皮肤病,包括三个主要类型(单纯型EB、交界型EB、营养不良型EB)和一种罕见变异型(Kinder综合征,即肢端角化性皮肤异色病),共分为至少20种临床亚型。角蛋白5和7、层粘连蛋-332、Ⅶ型胶原、大疱性类天疱疮抗原2、网格蛋白、整合素等7个靶蛋白中任何一个基因突变均可导致一种EB的发生。目前为止,已经发现的可导致EB发生的基因有19个,其中编码Ⅶ型胶原的COL7A1基因突变导致营养不良型EB的发生[2]。营养不良型EB分为常染色体显性遗传和常染色体隐性遗传,显性营养不良型EB(dominant dystrophic epidermolysis bullosa,DDEB)是由于Ⅶ型胶原蛋白发生负显性突变,突变点位于蛋白中糖基结合点,并不导致Ⅶ型胶原蛋白的截断,只是导致皮肤结构的异常,比如皮肤脆性增加、锚原纤维数量异常等。相反,隐性营养不良型EB(recessive dystrophic epidermolysis bullosa,RDEB)是Ⅶ型胶原蛋白的复合杂合子突变,提前出现的终止密码子导致蛋白的截断,从而导致Ⅶ型胶原的合成不稳定。从分子水平上看,本患者为编码Ⅶ型胶原的COL7A1基因外显子86上的错义点突变,没有导致Ⅶ型胶原蛋白的截断,家系遗传图谱也显示为常染色体显性遗传,符合DDEB的诊断。
与RDEB相比,DDEB一般临床表现较轻微[3],多在婴儿早期或儿童期发病,水疱及大疱多位于四肢伸侧,尤其是关节部位,尼氏征常为阴性,愈后形成粟丘疹和瘢痕。因为是蛋白合成异常所致,不是自身抗体的原因,因此免疫荧光是阴性的。DDEB分为以下亚型:肢端型、胫前型、痒疹型、仅指甲受累型及新生儿大疱性表皮松解型,其变异型包括白色丘疹样型和增生型。其中痒疹型DDEB表现为剧烈瘙痒性的紫红色丘疹和结节,轻度水疱或糜烂,呈线状排列,主要发生在胫前、前臂,少数发生在躯干,以及趾甲的萎缩变性,婴儿或儿童期发病。与本例两姐妹均在10岁左右发病,皮疹分布部位和皮疹特点都是相符的。结合临床、病理(表皮下裂隙,免疫荧光阴性)和分子遗传特点,符合痒疹型DDEB。
Masunaga等[4]总结了DEB的突变位点,提出隐性病例的突变分布于从N端到C端的整个VII型胶原。而与隐性病例相比,显性病例的突变位于COL7A1基因的末端,主要为甘氨酸替代突变。本例患者基因检测显示为甘氨酸被谷氨酸替代,与文献报道相符。本文中先证者及其胞妹COL7A1基因存在第6761位碱基G→A突变,导致第2254位甘氨酸替代,从而影响Ⅶ型胶原三螺旋结构区Gly-X-Y重复序列,破坏了三螺旋结构的稳定性。Clin Var数据库记录有该位点信息(c.6761G>A;p.Gly2254Glu),并评估为致病性变异。目前为止,只有2例国外文献报道了位于外显子86上的甘氨酸替代突变,分别是c.6797G>T(p.Gly2266Val)和c.6752G>A(p.G2251E),前者纯合突变来自一个索马里家庭[5],后者杂合突变来自一个日本家庭[6]。经搜索PubMed和万方数据库,国外报道并进行基因检测的DDEB共54例(表1),其中44例为甘氨酸替代突变,并且44例甘氨酸替代突变中以位于外显子73最多(23例),外显子87次之(4例)。因此国外常见的DDEB点突变位于外显子73。国内报道并进行基因检测的DDEB共10例(表2),其中9例为甘氨酸替代突变,而9例甘氨酸替代突变中有4例位于73号外显子。国内外DDEB以位于COL7A1基因73号外显子的点突变最常见,本例突变位于86号外显子,国内尚未见报道。

表1 国外报道的DDEB突变位点

续表

表2 国内报道的DDEB突变位点
综上,我们报道来自广东-DEB家族的COL7AI基因外显子86号上的杂合突变:c.6761G>A;p.Gly2254Glu,这一点突变是国内外首次报道。
猜你喜欢
分子诊断与治疗杂志(2022年9期)2022-10-09
今日农业(2022年3期)2022-06-05
中国农学通报(2022年6期)2022-04-15
医学前沿(2021年6期)2021-09-10
家庭科学·新健康(2020年6期)2020-07-06
祝您健康·文摘版(2019年4期)2019-06-11
中国美容医学(2019年2期)2019-03-13
饮食科学(2016年3期)2016-07-04
湖北农业科学(2014年11期)2014-09-10
中国针灸(2000年9期)2000-06-13
