
复发性线状棘层松解性皮病一例
2022-07-13 13:59贾凤铭刘永霞卢宪梅
中国麻风皮肤病杂志 2022年9期
贾凤铭 刘永霞 杨 青 卢宪梅
1 山东第一医科大学附属皮肤病医院( 山东省皮肤病医院),山东省皮肤病性病防治研究所,济南,250022; 2山东第一医科大学(山东省医学科学院),山东济南,250117
临床资料患儿,女,6岁。因臀部、右下肢屈侧条索状斑块反复出现5年余,加重2个月就诊。患者自1岁起无明显诱因于右侧臀部及右下肢屈侧出现暗红色斑块,境界较清晰,上覆鳞屑与结痂。皮损呈与肢体长轴平行的条索状排列,伴有轻度瘙痒。皮损出现后一段时间内能自行消退,但于同一位置反复出现,且近2个月皮损扩大、增多。病情严重时出现糜烂、渗出。曾以外用药膏治疗,具体药物名称及疗效不详。体格检查:患儿一般情况良好,系统体格检查未见异常。皮肤科检查:右侧臀部至右大腿屈侧及腘窝可见沿Blashko线分布的条索状暗红色斑块,边界清晰,上覆血痂和黄白色鳞屑,痂皮脱落处隐约可见红色糜烂面,未见明显渗出。散在色素沉着斑(图1),其余皮肤未见异常。辅助检查未见异常。否认既往系统性疾病病史及其他皮肤病病史,否认家族中有类似患者。
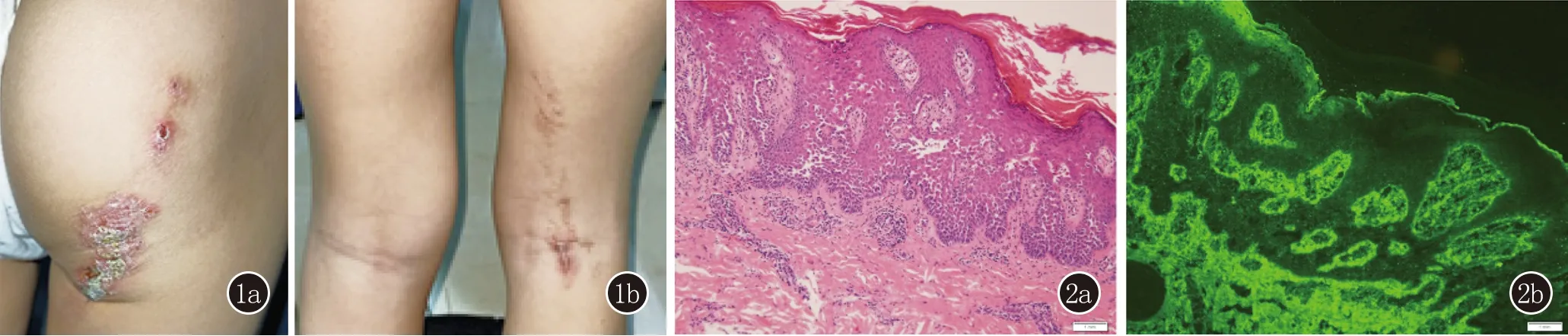
图1 1a、1b:右侧臀部至右大腿屈侧及腘窝可见沿Blashko线分布的条索状暗红色斑块,边界清晰,上覆血痂和黄白色鳞屑,痂皮脱落处隐约可见红色糜烂面 图2 2a:表皮角化过度、角化不全,棘层上方局灶嗜酸粒细胞脓肿,棘层增厚,棘层松解呈砖墙倒塌样,个别嗜酸粒细胞移入,真皮浅层血管周围少许淋巴细胞、个别嗜酸粒细胞浸润(HE,×100);2b:DIF见表皮细胞及基底膜IgG、C3、IgM、IgA阴性 (直接免疫荧光,×100)
取右侧臀部皮损行组织病理学检查结果示表皮角化过度、角化不全,棘层上方局灶嗜酸粒细胞脓肿,棘层增厚,棘层松解呈砖墙倒塌样,个别嗜酸粒细胞移入,真皮浅层血管周围少许淋巴细胞、个别嗜酸粒细胞浸润。DIF见表皮细胞及基底膜IgG、C3、IgM、IgA阴性(图2)。
诊断:复发性线状棘层松解性皮病。治疗:给予复方硝酸咪康唑乳膏日2次外用,两周后随访,皮损有明显好转。治疗2月后随访,皮损较前无明显变化,臀部见增生性瘢痕,腘窝有棕色色素沉着(图3)。

3a:臀部皮损;3b:腘窝皮损
讨论复发性线状棘层松解性皮病(relapsing linear acantholytic dermatosis)是一种比较少见的,组织病理表现与家族性良性慢性天疱疮(Hailey-Hailey病,HHD)相似的表皮痣样疾病,又被称为Hailey-Hailey样表皮痣。本病由Vakilzadeh和Kolde在1985年首报[1],该病皮损特点为沿Blaschko线分布的红斑、斑块,上覆群集的水疱,水疱破溃后形成表浅的糜烂,结痂。皮损可自行缓解,但病情反复。组织病理特点为基底层上裂隙形成和表皮内大面积出现部分性或完全性的棘层松解,棘层松解呈塌砖墙样外观。成熟皮损内可见水疱和大疱形成,单层基底细胞的乳突向上突入间隙内。直接、间接免疫荧光染色阴性。既往报道本病7例,临床特征与组织病理学表现见表1。
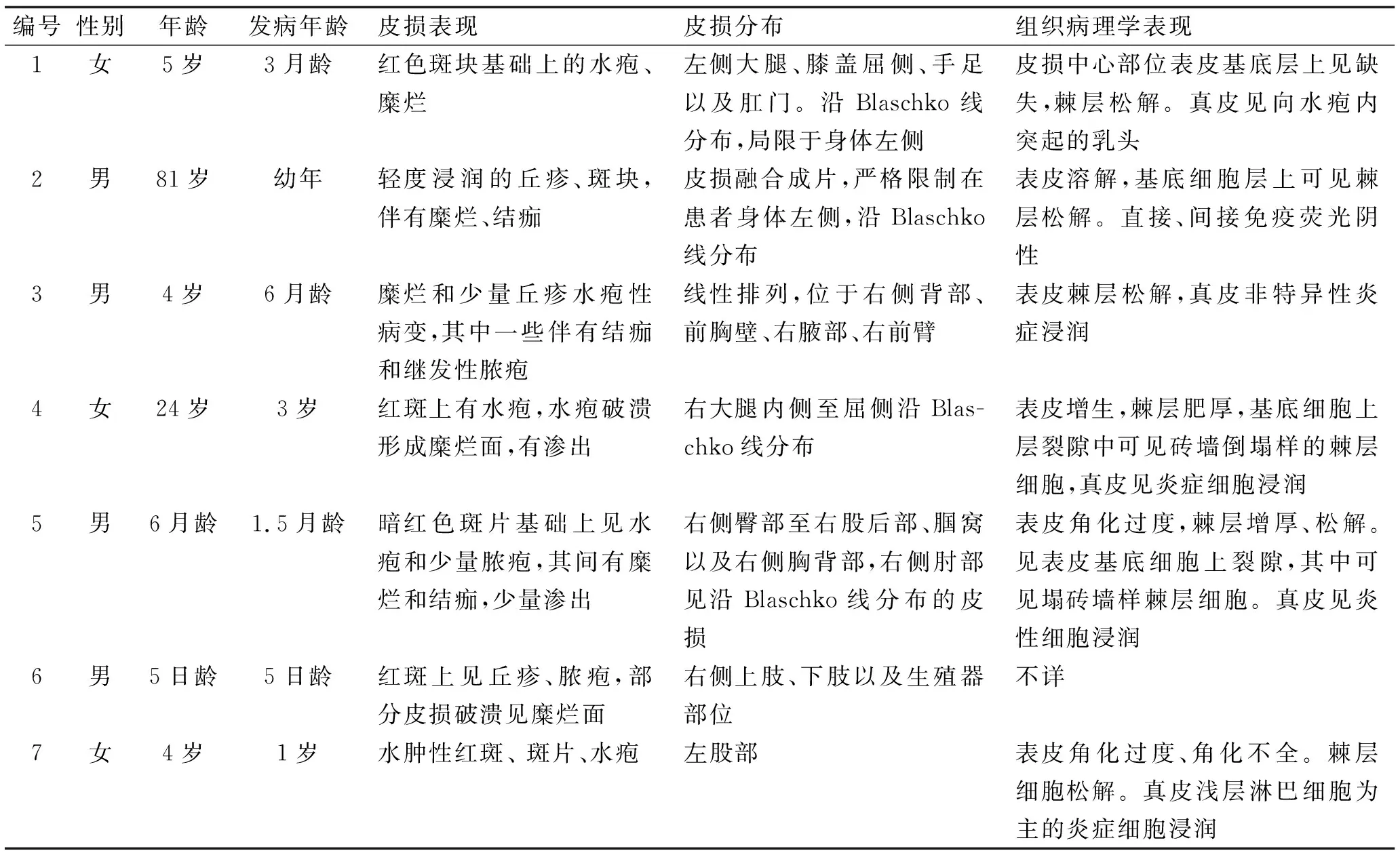
表1 7例复发性棘层松解性皮病[1-7]
由于起病年龄、皮损分布、是否累计掌跖等方面存在明显区别,在Vakilzadeh[8]1985年的首报中,本病被认为是一种独立的棘层松解性疾病。上世纪七十年代,多种疾病被证实是由于基因突变与正常胚胎的镶嵌体导致,这种受精卵形成后的突变使得携带突变的嵌合体胚胎能够存活并表现出交替分布的节段性性状,沿Blaschko线分布的皮损表现被认为是嵌合体的皮肤表现之一。Katzman等[9]2020年在复发性线状棘层松解性皮病的皮损中发现多个ATP2C1基因突变位点,而上述突变不存在于病人的正常皮肤以及外周血DNA中,证实该突变可能是来源于受精卵形成后某时期,获得该突变的某胚胎原始细胞不断增殖、分化、迁延,形成了沿Blaschko线的突变细胞分布形态。ATP2C1基因位于3号染色体上,编码一种钙离子转运体蛋白,主要负责高尔基体对钙离子、锰离子的摄取。2000年,Hu等[10]首次报道了ATP2C1基因突变能够导致HHD的发生。日本,中国等多个HHD家系中也陆续发现了多种ATP2C1基因的突变[11-13]。在复发性线状棘层松解性皮病发现的ATP2C1基因突变一定程度上支持了本病是Hailey-Hailey病的一种特殊临床类型这一观点。
除了Hailey-Hailey病,本病还应与局限性Darier病、带状分布的Grover病、线状疣状表皮痣以及多种免疫大疱性皮肤病相鉴别。Darier病又称为毛囊角化病,与ATP2A2基因突变相关,常于中青年起病,典型部位为皮脂溢出部位,皮损为细小坚实的皮色丘疹,丘疹去除后可见漏斗样小凹,很少出现水疱、糜烂、渗出、结痂等皮损表现。局限性Darier病皮损呈局限性或沿带状分布,夏季症状常见加重,组织病理学表现为特殊形态的角化不良,形成圆体和谷粒,基底层上棘层松解,真皮呈慢性炎症性浸润。Grover病又名暂时性棘层松解性皮病,皮损为散发的水肿型丘疹或丘疱疹,组织病理特征为棘层肥厚,表皮内见局灶性棘层松解,表皮内可见海绵形成。线状疣状表皮痣多自幼年起病,皮损表现为淡黄色至棕黑色疣状损害,可位于身体任何部位,也可呈线状或仅在身体单侧分布。病变进展缓慢,无周期性缓解,一般无自觉症状。免疫性大疱性皮病可通过病史以及特异性抗体检测和DIF、IIF等组织病理学表现予以鉴别。
本例患者自幼年起病,病情反复,迁延不愈。皮损自臀部、股后部至右下肢呈条索状排列,符合Blaschko线在下肢的走向特点。组织病理学检查示砖墙倒塌样的棘层松解,DIF阴性。结合临床表现以及组织病理学特点,符合该病诊断。相比既往报道的病例,本例患儿起病症状较轻且不典型,皮损局限性分布在臀部和股部区域且无明显的水疱、脓疱、糜烂,未见明显渗出,上覆少许鳞屑和干燥的结痂,临床上易与线状苔藓、线状扁平苔藓、线状银屑病等呈线状排列的红斑鳞屑性皮肤病混淆,从而忽视本病的可能性,误导治疗方向。针对皮损表现较轻的本病患者,嘱皮损部位避光,外用糖皮质激素以及抗生素制剂可以获得较为满意的皮损改善;针对皮损泛发,融合成片且合并较为严重的水疱、脓疱及渗出,外用药物控制不佳者,可以尝试参考HHD患者的治疗方案:如系统使用抗生素和糖皮质激素、环孢素、维A酸和氨苯砜等。鉴于本病具有自发性缓解周期,有必要对患者进行长期随访,观察后续复发以及缓解情况。
猜你喜欢
纺织高校基础科学学报(2022年2期)2022-07-29
临床骨科杂志(2022年3期)2022-06-24
中国典型病例大全(2022年9期)2022-04-19
数学学习与研究(2021年18期)2021-08-06
中国药学药品知识仓库(2021年18期)2021-02-28
银潮(2020年10期)2020-10-23
爱你·健康读本(2019年8期)2019-11-22
爱你(2019年29期)2019-11-07
环球市场信息导报(2018年21期)2018-07-27
东方教育(2017年14期)2017-09-25
