
Escherichia coli Protoplast Fusion and Screening of High L-Lysine-Producing Strain
2022-07-13 01:43ZiruiWANGJunqingWANG
农业生物技术(英文版) 2022年3期
Zirui WANG Junqing WANG


Abstract[Objectives] Protoplast fusion of two parental strains with weak L-lysine production ability was carried out to obtain new fusion strains with strong L-lysine production ability. [Methods]The effects of bacterial age, lysozyme concentration, enzymolysis temperature and time on the protoplast formation rate and regeneration rate were investigated by single factor experiments. On this basis, with the protoplast formation rate as an index, the protoplast preparation process was optimized by an orthogonal experiment. [Results] Bacterial age and enzymolysis time had a greater impact on the protoplast formation rate, followed by enzymolysis temperature and lysozyme concentration. The optimal process for the preparation of L-lysine-producing Escherichia coli protoplasts was to prepare parental protoplasts from bacterial cells cultured for 15 h in the late logarithmic growth phase by enzymolysis with 0.8 mg/ml lysozyme at 37 ℃ for 180 min and promote fusion with PEG6000. In order to facilitate the screening of fusion protoplasts, the empty plasmids pET-28a and pET-Duet were transformed into L-lysine-producing E. coli, respectively, and strains pET-28a-lys01 and pET-Duet-lys01 were obtained. Fusion strains were then obtained through protoplast fusion. Double-resistance KA1-10 were screened on plates containing kanamycin and ampicillin, and a high-yielding fusion strain KA8, which produced L-lysine, was screened by fermentation experiments finally. [Conclusions]The results of this study provide a reference method for further improving the yield of L-lysine and other amino acid strains.
Key wordsEscherichia coli; L-lysine; Protoplasts; Cell fusion; Microbial breeding
Protoplast fusion, also known as cell fusion, is a technology in which the genetic material of one cell, including DNA and extranuclear genes, enters another cell[1]. Its purpose is to make the genetic gene of another cell entering the cell to be accepted by the cell, or to cause the variation of the genetic characteristics of the cell, and such accepted genetic characteristics or genetic variation that occurs can be passed down and stabilized from generation to generation. At present, protoplast fusion has been widely used in animal and plant breeding, and great progress has been made. In the field of microbiology, there are also many successful examples. For example, some people have made protoplast fusion between the varieties of wuyiencin and povamycin-producing bacteria, and applied it to agricultural antibiotics and the selection and breeding of producing bacteria, and achieved success. For another example, the fusion of Brevibacterium cells developed by Ajinomoto, a Japanese company, can cultivate production strains that simultaneously produce L-threonine and L-lysine. Mutant strain PF-25 produced by cell fusion of erythromycin-producing bacteria and tetracycline-producing bacteria can greatly increase the production of daunorubicin[2]. Pang et al.[3] used alcoholic yeast and Candida tropicalis as parents, and obtained a stable yeast fusion strain with both saccharifying enzyme activity and high alcohol production through protoplast fusion technique, and the alcohol fermentation yield could reach 8.8% and 11.5%. Gao et al.[4] carried out intergeneric protoplast fusion selection between Saccharomyces cerevisiae and Schizosaccharomyces pombe to obtain a strain with strong malic acid degradation. Practice has proved that the use of protoplast fusion technology to optimize different characteristics of different species of microorganisms to produce new varieties can not only avoid the disadvantages of high investment and poor effect of some physical and chemical methods, but also effectively reduce pollution and benefit mankind more efficiently.
The chemical name of L-lysine is 2,6-diaminocaproic acid, and the molecular formula is CHNO[5]. There are three optical isomers: L-form (left-handed), D-form (right-handed) and DL-form (racemic), but only the L-form can be absorbed and utilized by humans and animals[6]. L-Lysine is one of the amino acids necessary for humans and animals that cannot be synthesized by itself. It is widely used in feed additives, food fortifiers and pharmaceutical products. More than 90% of L-lysine products are used as feed additives[7]. Studies have shown that adding an appropriate amount of L-lysine to grain feed can greatly improve the utilization rate of protein and promote the growth of livestock. L-lysine can determine protein synthesis and deposition and the utilization of other amino acids in weaned piglets, and the conversion efficiency of L-lysine into body protein is as high as 86%[8]. At present, the production of lysine at home and abroad adopts the fermentation method. The acid production rate of the glutamic acid-producing bacteria selected by conventional breeding technology is about 13% to 17%, and the fermentation period is 60 to 64 h. The acid production rate of the strains transformed from Escherichia coli by genetic engineering technology is 16%-19%, and the fermentation period is 40-50 h[9-10]. The synthesis of lysine in microorganisms and plants has two completely different routes: the diaminopimelate pathway and the α-aminoadipic acid pathway[11-12]. The microorganisms used for the production of L-lysine by fermentation are Corynebacterium glutamicum, E. coli, Brevibacterium flavum, S. cerevisiae, Brevibacterium lactofermentum, Candida, Corynebacterium acetoacidophilum and Corynebacterium thermoaminogenes, etc.[13-16]. Protoplast fusion is a key step in obtaining stable fusants in microbial fusion breeding technology, mainly through a series of methods for recombining genes[17]. The essence of protoplast preparation is to break the wall to free protoplasts, and regeneration is to regenerate the lost cell wall. The two processes are separate but closely related[18]. Bacteria and actinomycetes have a similar cell wall component, peptidoglycan, so the same method is often used to prepare protoplasts, that is, lysozyme is used as a tool enzyme[19-20]. China's demand for lysine is at the forefront of all countries in the world. In the study, a new fusion strain with strong L-lysine production ability was obtained by protoplast fusion of two parental strains with low L-lysine production ability. After the fusion of protoplasts, genomes of the two strains undergone a series of recombination changes such as insertion and exchange, so that the ability of the strain to produce L-lysine was improved.
Materials and Methods
Strains and plasmids
In the research process, we selected empty plasmid pET-28a and empty plasmid pET-Duet, which were transformed using E. coli DH5α as the strain for plasmid preservation and preparation and E. coli QD01 as the host, respectively. Protoplast fusion was carried out based on different resistance of the plasmids. The plasmids and strains used are shown in Table 1.
Reagents and mediums
Protoplast stabilization solution (SMM): a pH of 6.5, 0.5 mol/L sucrose, 20 mmol/L anhydrous magnesium chloride and 0.02 mol/L maleic acid; PEG6000 liquid (40%): 40% PEG6000 was added to the protoplast stabilization solution (pH=6.5); lysozyme liquid: lysozyme liquid with concentrations of 0.2, 0.4, 0.6, 0.8, 1.0 and 1.2 mg/ml were prepared, respectively, and filtered through a 0.22 μm filter membranes, and after aliquoting (1 ml/tube), they were stored at -20 ℃; kanamycin antibiotic (50 mg/ml): 0.5 g of kanamycin was weighed into a 10 ml plastic centrifuge tube and added with 8 ml of sterilized water, and after fully mixing and dissolution, the solution was diluted to 10 ml and filtered through a 0.22 μm filter membrane, obtaining a filtrate, which was aliquoted into small portions (1 ml/tube), which were stored at -20 ℃; ampicillin (100 mg/ml): 1 g of ampicillin was weighed to a 10 ml plastic centrifuge tube and added with 8 ml of sterile water, and after fully mixing and dissolution, and the solution was diluted to 10 ml and filtered through a 0.22 μm filter membrane, obtaining a filtrate, which was aliquoted into small portions (1 ml/tube), which were stored at -20 ℃; fresh calcium phosphate solution: 0.54 g of dipotassium hydrogen phosphate was weighed and dissolved in 100 ml of water, and 29.4 g of anhydrous calcium chloride was weighed and dissolved in 100 ml of water.
LB solid medium: tryptone 1%, yeast extract 0.5%, sodium chloride 10%, agar powder 2%; LB liquid medium: the formula was the same as LB medium, without the addition of agar.
Strain preparation and screening method
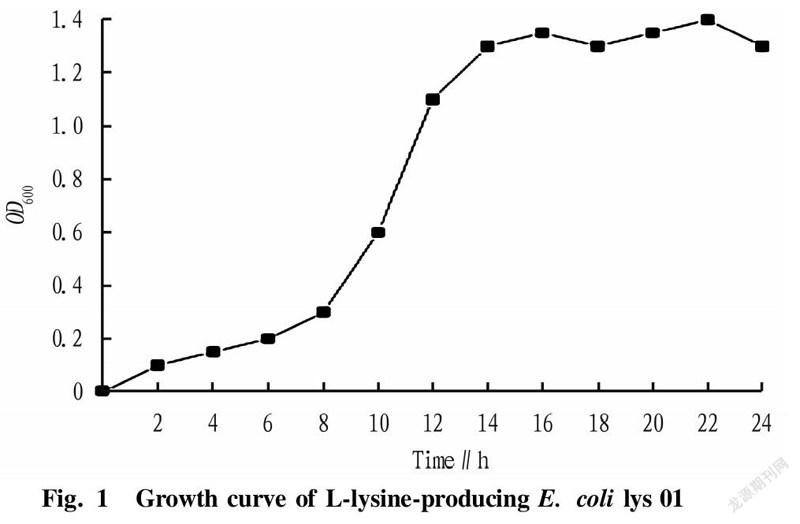
Screening of the best bacterial ageThe OD value of microorganisms is an index reflecting the growth state of bacteria. OD is the abbreviation of optical density, which represents the optical density absorbed by the detected substance. Bacteria are usually detected at 600 nm. L-lysine-producing starting strain lys 01 was streak-inoculated into shake flasks for strain growth curve measurement, and measurement was performed once every 2 h. The growth curve of L-lysine-producing E. coli lys 01 within 24 h was measured. According to different growth rates, the growth curve could generally be divided into the delayed phase, logarithmic phase, stable phase and decline phase. The lengths of these four phases vary with the heredity, inoculum amount and culture conditions of strains. After comparing the recovery rate, we knew that the phase with the highest recovery rate of protoplasts was the late logarithmic growth phase.
Screening of optimal enzyme concentrationThe two strains were inoculated into LB medium at an amount of 1%, and cultured at 37 ℃and 200 r/min to mid-logarithmic phase. Next, 10 ml of bacterial liquid was collected and washed for the bacterial cells with sterile water twice. The bacterial cells were finally adjusted to the bacterial concentration of 1×107 CFU/ml with SMM buffer solution. Lysozyme with concentrations of 0.2, 0.4, 0.6, 0.8, 1.0 and 1.2 mg/ml was added, and enzymatic hydrolysis was carried out at 37 ℃ for 30 min. After the enzymatic hydrolysis was completed, the cells were washed twice immediately. Each bacterial liquid was diluted to 1×10-5 with sterile water and SMM solution, respectively, and after standing for 30 min, 0.1 ml of each bacterial liquid was coated on LB medium.
Screening of optimal enzymatic hydrolysis timeThe concentrations of the two parental strains were adjusted to 1×10 CFU/ml in the same way as above. Under the condition of the optimal enzymatic hydrolysis concentration, different enzymatic hydrolysis time was set respectively to explore the effect of time on protoplast formation of the parental strains. The bacterial liquid after enzymolysis was immediately centrifuged and the bacterial cells were washed twice. Each bacterial liquid was diluted to 1×10 with sterile water and SMM solution, respectively, and 0.1 ml of each bacterial liquid was coated on LB medium.
Calculation of protoplast formation rate and regeneration rate:
Protoplast formation rate=(A-B)/A×100%
Regeneration rate=(C-B)/(A-B)×100%
In the formulas, A: the number of colonies on the LB plate before lysozyme treatment; B: the number of colonies without protoplastization after lysozyme treatment; C: the number of colonies on the LB plate after lysozyme treatment.
A certain volume of E. coli protoplast liquid was diluted and counted under a microscope. Equal volumes of the above two kinds of protoplasts were washed with SMM buffer, coated on LB medium, and incubated at 28 ℃ for 2-3 d. The number of colonies formed on LB medium after the protoplast liquids were diluted with SMM buffer was observed, and the protoplast regeneration rate was calculated according to protoplast regeneration rate=(A-B)C×100% (A=Number of colonies formed after dilution with SMM buffer). In protoplast fusion, the same amount of parental protoplast liquids (the number of protoplasts≥107 ml) diluted with SMM buffer were mixed, and the obtained mixture was centrifuged at 3 000 r/min in for 10 min. After discarding the supernatant, the tube wall was tapped with fingers to make the precipitated protoplasts dispersed in the remaining droplets. Next, 5 ml of SMM buffer was added to wash once, and then the protoplasts were re-dispersed in the remaining droplets. Next, 1 to 2 ml of 40% PEG solution prepared in SMM buffer was added, and the liquid was mixed uniformly by immediately pipetted once. The liquid was placed at 30-37 ℃ for 5 min (the fusion process was photographed with a phase contrast microscope), diluted with 5 ml of SMM buffer, and centrifuged at 3 000 rpm, and the supernatant was removed. The precipitate was uniformly suspended in a 2 ml of buffer solution, and 0.1 ml of the protoplast liquid was spread on LB medium and incubated at 28 ℃ for 2-3 d.
Preparation of E. coli lys 01 competent cellsFirst, a single colony was picked with a pipette tip, put into a 50 ml test tube containing 10 ml of LB liquid medium, and incubated at 37 ℃ and 220 r/min for 14-16 h. Then, the bacteria were inoculated with an inoculation quantity of 1% to 1 000 ml LB liquid medium, and the obtained liquid was shaken at 220 r/min and 37 ℃ for 2-3 h. The culture was stopped when the OD6 value reached 0.3-0.4. Then, the bacterial liquid was pre-cooled on ice for half an hour, and was dispensed into 500 ml pre-cooled centrifuge cups, and centrifuged at 2 500 r/min at 4 ℃ for 10 min. Then, the supernatant was discarded, and the precipitate was suspended by adding a small amount of ddHO to each centrifuge cup, which was then filled with water. Each obtained liquid was centrifuged at 4 000 r/min for 10 min at 4 ℃. Next, after discarding the supernatant, the cells were added with a small amount of sterile water to re-suspend the bacteria, and each centrifuge cup was then filled with water. Each suspension was centrifuged at 4 000 r/min for 10 min at 4 ℃. Then, the supernatant was discarded, and a small amount of 10% glycerol was added to each centrifuge cup to re-suspend the cells. Each centrifuge cup was filled with 10% glycerol to perform centrifuge at 4 000 r/min at 4 ℃ for 10 min. Finally, the supernatant was discarded, 5 ml of 10% glycerol was added to each centrifuge cup to suspend the precipitate, and the obtained bacterial liquid was added into 1.5 ml centrifuge tubes at 100 μl/tube and stored in a -80 ℃ refrigerator.
Transformation of plasmid pET-28a-lys 01 and plasmid pET-Duet-lys 01Plasmid pET-28a and plasmid pET-Duet were transferred into lys 01 competent cells, respectively. Before the experiment, the water baths used were adjusted to 42 ℃ and heated to the preset temperature in advance. Then, lys 01 competent cells were thawed on ice, and 1 μl of 1 μg/μl plasmid was added to 10 μl of competent cells, obtaining a mixture, which was placed on ice for 30 min. Each mixture was subjected to heat shock in a water bath at 42 ℃ for 90 s and quickly transferred to ice for 2 to 5 min. In an aseptic bench, 200 μl of LB medium (no resistance) was added to each competent cell tube containing corresponding plasmid, and the cells were then transferred to culture tubes and cultured at a low speed for 1 h at 37 ℃. Next, a 200 μl of suspension was poured onto corresponding resistant LB solid medium, and a coating rod was sterilized on an alcohol lamp and used to spread the suspension evenly. The bacteria were then cultured upside down in a constant temperature incubator at 37 ℃ for 12-16 h, and the growth of colonies was observed the next day. The two plasmids were respectively transferred into lys 01 competent cells, and the specific transformation steps were carried out according to the above steps. The whole process should be carried out on ice. After taking out the competent cells from a -80 ℃ refrigerator, it should be quickly placed on ice to melt it, and the prepared pET-28a and pET-duet plasmids were transformed according to actual situations. After transformation and recovery, 200 μl of re-suspended bacteria was left by centrifugation for coating. After overnight culture, the cells were picked for resistance verification on the second day. K-lys 01 and A-lys 01 strains were obtained respectively.
Protoplast fusionFirst, 0.5 ml of the above-mentioned parental strains K-lys 01 and A-lys 01 were mixed and centrifuged for 5 min (2 500 r/min, 10 min) to collect the protoplasts. The collected protoplasts were added with 4.8 ml of 40% PEG6000 solution and 0.2 ml of fresh calcium phosphate solution, and the liquid was mixed uniformly by gently pipetting, and then incubated at 30 ℃ for 15-30 min. The fusion process was observed with a microscope. Centrifugation was then performed at 2 500 r/min for 10 min, and the precipitate was washed once with SMM buffer and then re-suspended with 0.5 ml of SMM buffer. Next, 100 μl was coated to a double-antibiotic regeneration medium plate, and incubated at 37 ℃ for 3-5 d (if it was stored for a long time, it would be put upside down and added with water). After the observed colonies were picked to the seed medium, another 100 μl was added to the double-antibiotic regeneration medium and directly cultivated at 37 ℃, and then streak inoculation was performed for colony identification.
Picking was performed on regeneration plates containing kanamycin and ampicillin. The above strains were added to a 96-well plate containing LB medium with kanamycin and ampicillin resistance for 48 h of growth. The above-mentioned fast-growing fusion strains were then transferred to a fermentation medium for fermentation flask experiments, and samples were taken to measure the L-lysine production by an SBA biosensor analyzer, so as to obtain double-resistance E. coli fusion strains with high L-lysine production.
Electron microscope morphological analysis of fusion strainIn this study, light microscopy was used to observe the sizes of the fusion strain and the original strains for comparison. The fusant stability analysis is that fusion strains are prone to degeneration after multiple passages, resulting in the strains returning to their previous states. After fusion strains KA1 to KA8 were passaged for 5 generations, the stability of the fusion strains was verified, and the stability of the passaged fusion strains was analyzed by fermentation experiments. Fusion strain KA8 with stable growth rate and stable acid production was selected.
Results and Discussion
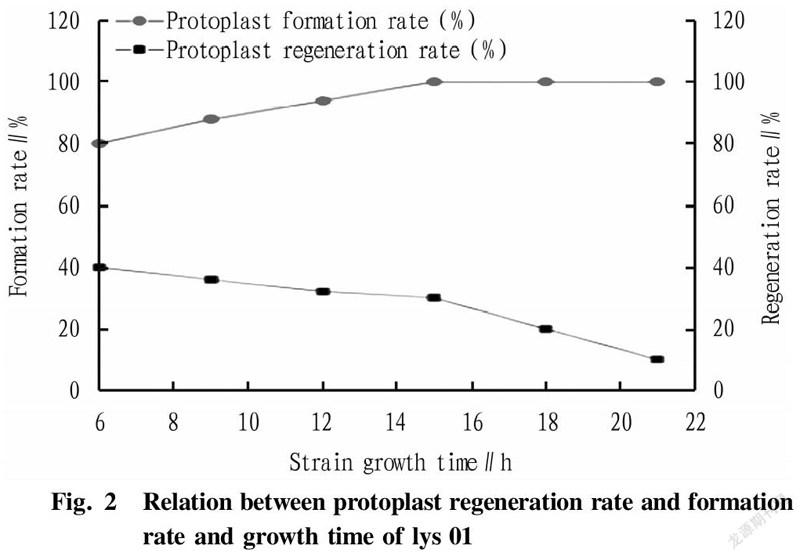
In the research experiment, the L-lysine-producing starting strain lys 01 was streak-inoculated into shake flasks to measure the growth curve of the strain. The growth curve of L-lysine-producing E. coli lys 01 was drawn over 24 h by measuring every 2 h (Fig. 1). According to the different growth rate, the growth curve could generally be divided into the delayed phase, logarithmic phase, stable phase and decline phase. The length of these four phases varies with the heredity, inoculum amount and culture conditions of strains. Maintaining a single variable in the study, it could be concluded from Fig. 1 and Fig. 2 that the phase with the highest recovery rate of protoplasts was 15 h (late logarithmic growth phase).
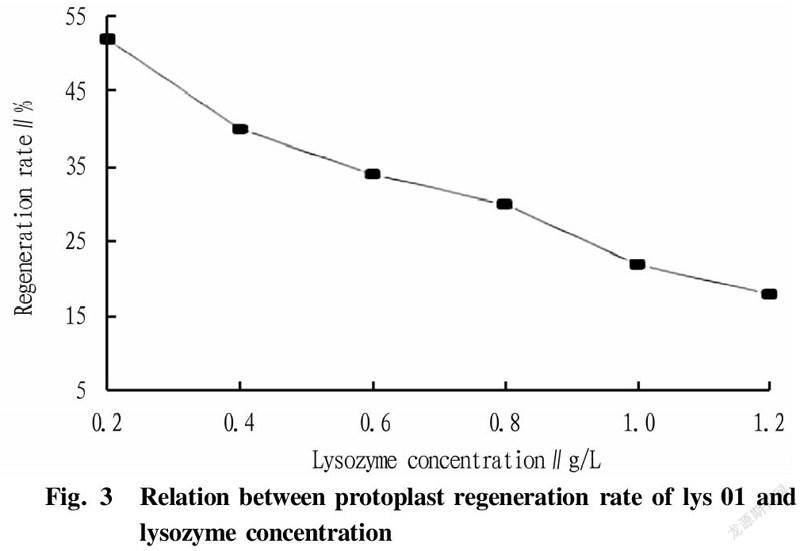
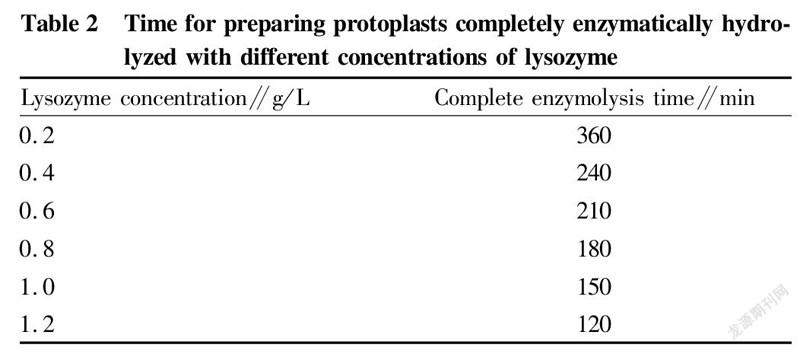
The two strains were inoculated into LB medium at an inoculation quantity of 1%, and cultured at 37 ℃ and 200 r/min to the mid-logarithmic phase. Then, 10 ml of each bacterial liquid was collected, washed twice with sterile water, and finally adjusted to 1×10 CFU/ml with SMM buffer solution. Lysozyme with concentrations of 0.2, 0.4, 0.6, 0.8, 1.0, and 1.2 mg/ml was added, and the cells were hydrolyzed at 37 ℃ for 30 min. The cells were washed twice immediately after the enzymatic hydrolysis was completed, and diluted to 1×10with sterile water and SMM buffer solution, respectively. After standing for 30 min, 0.1 ml of bacterial liquid was spread on LB medium. According to Fig. 3, it can be concluded that the optimal enzyme concentration was 0.8 mg/ml. According to the experimental data in Table 2 and Fig. 3, it was concluded that when the bacterial cells were hydrolyzed with 0.8 mg/ml lysozyme at 37 ℃ for 180 min, the formation efficiency of protoplast recovery (regeneration rate/complete enzymolysis time) was the highest.
In the study, E. coli lys 01 competent cells were prepared first. First, a single colony was picked with a pipette tip, put into a 50 ml test tube containing 10 ml of LB liquid medium, and cultured at 37 ℃ and 220 r/min for 14-16 h. Then, the bacteria were inoculated at an inoculation quantity of 1% to 1 000 ml of LB liquid medium, which was shaken for 2-3 h at 37 ℃ and 220 r/min. The culture was stopped when the OD600 value reached 0.3-0.4. Next, the bacterial liquid was pre-cooled on ice for half an hour, and was dispensed into 500 ml pre-cooled centrifuge cups, and centrifuged at 2 500 r/min at 4 ℃ for 10 min. Then, the supernatant was discarded, and the precipitate was suspended by adding a small amount of ddH2O to each centrifuge cup, which was then filled with water. Each obtained liquid was centrifuged at 4 000 r/min for 10 min at 4 ℃. Next, after discarding the supernatant, the cells were added with a small amount of sterile water to re-suspend the bacteria, and each centrifuge cup was then filled with water. Each suspension was centrifuged at 4 000 r/min for 10 min at 4 ℃. Then, the supernatant was discarded, and a small amount of 10% glycerol was added to each centrifuge cup to re-suspend the cells. Each centrifuge cup was filled with 10% glycerol to perform centrifuge at 4 000 r/min at 4 ℃ for 10 min. Finally, the supernatant was discarded, 5 ml of 10% glycerol was added to each centrifuge cup to suspend the precipitate. Plasmid pET-28a and plasmid pET-Duet were transferred into lys 01 competent cells, respectively. Before the experiment, the water baths used were adjusted to 42 ℃ and heated to the preset temperature in advance. Then, lys 01 competent cells were thawed on ice, and 1 μl of 1 μg/μl plasmid was added to 10 μl of competent cells, obtaining a mixture, which was placed on ice for 30 min. Each mixture was subjected to heat shock in a water bath at 42 ℃ for 90 s and quickly transferred to ice for 2 to 5 min. In an aseptic bench, 200 μl of LB medium (no resistance) was added to each competent cell tube containing corresponding plasmid, and the cells were then transferred to culture tubes and cultured at a low speed for 1 h at 37 ℃. Next, a 200 μl of suspension was poured onto corresponding resistant LB solid medium, and a coating rod was sterilized on an alcohol lamp and used to spread the suspension evenly. The bacteria were then cultured upside down in a constant temperature incubator at 37 ℃ for 12-16 h, and the growth of colonies was observed the next day. The two plasmids were respectively transferred into lys 01 competent cells, and the specific transformation steps were carried out according to the above steps. The whole process should be carried out on ice. After taking out the competent cells from a -80 ℃ refrigerator, it should be quickly placed on ice to melt it, and the prepared pET-28a and pET-duet plasmids were transformed according to actual situations. After transformation and recovery, 200 μl of re-suspended bacteria was left by centrifugation for coating. After overnight culture, the cells were picked for resistance verification on the second day. K-lys 01 and A-lys 01 strains were obtained respectively.
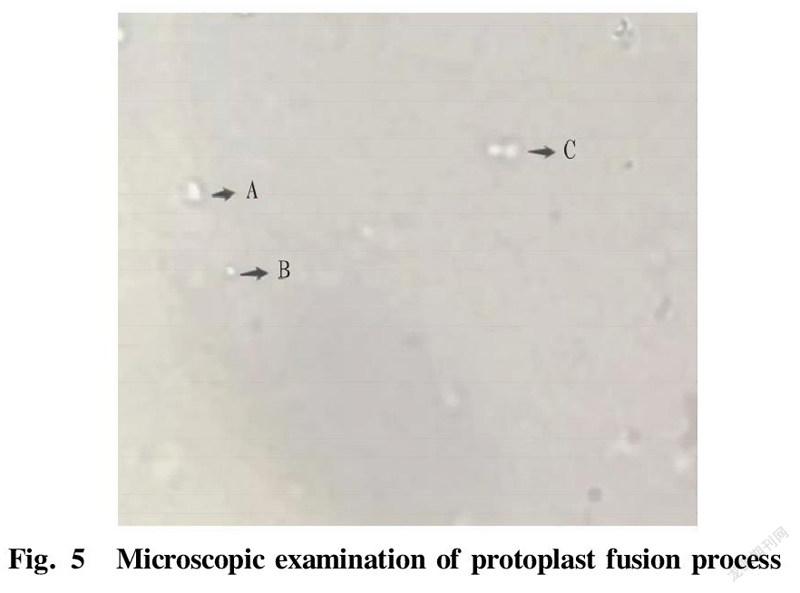
In protoplast funsion, 0.5 ml of the above-mentioned parental strains K-lys 01 and A-lys 01 were mixed and centrifuged for 5 min (2 500 r/min, 10 min) to collect the protoplasts. The collected protoplasts were added with 4.8 ml of 40% PEG6000 solution and 0.2 ml of fresh calcium phosphate solution, and the liquid was mixed uniformly by gently pipetting, and then incubated at 30 ℃ for 15-30 min. The fusion process was observed with a microscope. Centrifugation was then performed at 2 500 r/min for 10 min, and the precipitate was washed once with SMM buffer and then re-suspended with 0.5 ml of SMM buffer. Next, 100 μl was coated to a double-antibiotic regeneration medium plate, and incubated at 37 ℃. After the observed colonies were picked to the seed medium, another 100 μl was added to the double-antibiotic regeneration medium and directly cultivated at 37 ℃, and then streak inoculation was performed for colony identification. Picking was performed on regeneration plates containing kanamycin and ampicillin. The above strains were added to a 96-well plate containing LB medium with kanamycin and ampicillin resistance for 48 h of growth. The above-mentioned fast-growing fusion strains were then transferred to a fermentation medium for fermentation flask experiments. The growth curves of three strains measured by sampling are shown in Fig. 4. The growth rates of the fusion strains were lower than that of the starting strain, because the genomes of the fusion strains were too large, resulting in a decrease in the growth rates of the fusion strains. According to the microscopic examination in Fig. 5, it can be seen that A is the state of a fusant after the fusion of two protoplasts; B is the state of a single protoplast; and C is the state of two protoplasts when they are close. In terms of size and shape, fusant A is larger than single protoplast B. Samples were taken and L-lysine production was measured by an SBA biosensor analyzer. A double-resistance E. coli fusant strain KA8 with high L-lysine production was obtained.
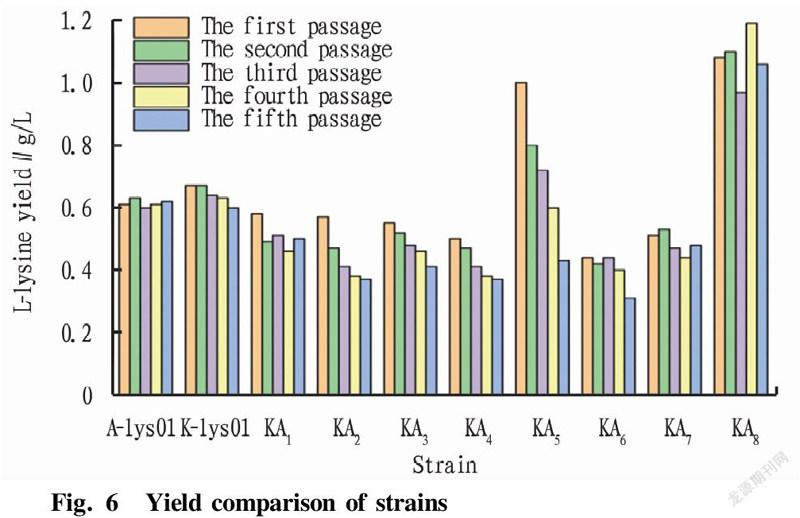
After the first round of passage, two strains, KA and KA, were screened out with no yield. The remaining eight strains were subjected to stability passage experiments. Each passage was fermented for 48 h, and samples were taken for L-lysine content determination using an SBA biosensor analyzer. According to Fig. 6, E. coli fusion strain KA with high L-lysine production was screened. The average L-lysine production of starting strains A-lys 01 and K-lys 01 was 0.62 g/L, and the average L-lysine production of the fusion strains KA, KA, KA, KA, KA and KA was lower than 0.6 g/L, so they were weeded out. The L-lysine fermentation yield of strain KA5 decreased gradually at each passage, that is, the fermentation yield was unstable, which might be caused by the loss of the L-lysine-producing gene during the passage of the fusion strain. After 5 passages of strain KA, the L-lysine yield during fermentation was relatively stable with an average L-lysine yield at about 1.0 g/L, and the L-lysine yield was 66.7% higher than that of the starting strain. Therefore, stable and high-yielding fusion strain KA was screened out. In this study, a new fusion strain KA with strong L-lysine production ability was obtained by protoplast fusion of two parental strains with low L-lysine production ability. After the fusion of the protoplasts, a series of recombination changes such as insertion and exchange occurred in the genomes of the two strains, so that the ability of the strain to produce L-lysine was improved.
Conclusions and Discussion
As a promising substance, L-lysine has been widely favored by scientists, and solving the current limitation of the ability of wild-type strains to produce L-lysine is also a key issue of research. As a traditional breeding technology, protoplast fusion has the advantages of combining the excellent characters of the two parental strains, and the operation is simple and convenient. In this study, a new fusion strain with strong L-lysine production ability was obtained by protoplast fusion of two parental strains with low L-lysine production ability. The reason might be that after the fusion of protoplasts and a series of recombination changes such as insertion and exchange of the genomes of the two strains, the ability of the strain to produce L-lysine was improved. For the screening of high L-lysine-producing strains, the main problem is how to quickly screen out target strains. In previous studies, the screening methods for high L-lysine-producing strains could not accurately screen out target strains, and the possible reason was that E. coli can produce many substances with similar properties to L-lysine, such as antibacterial proteins and glycolipids, all of which will greatly interfere with the screening results, resulting in increased screening errors. Compared with some other screening methods, the method of preparing fusants by resistance in this study can quickly screen out our target strains through resistance screening, which greatly saves screening time and workload. For fusion strain KA8 obtained by the final screening, the resistance screening was verified, and then shake flask fermentation was performed to measure the L-lysine-producing ability of the fusant, which was improved, indicating that the method can effectively high-producing L-lysine strains.
In this study, it was found that the method of screening fusion strains by 96-well plates, measuring strains with fast growth rate by a microplate reader and finally obtaining high-yielding L-lysine double-resistance fusants by fermentation culture using the double-antibiotic fermentation medium was simple and fast. In addition, studies have shown that fusion strains are unstable, and the fusion characteristics of strains will disappear after several passages. Fusion KA in this study could still produce L-lysine stably after more than 5 passages, indicating that the fusant was stable, and other 9 strains did not obtain high production of L-lysine, indicating that the fusion trait was lost. The electron microscope shape observation of the KA fusion strain also showed that the fusion strain has become larger in shape, which further proves that KA8 fused the parental genomes. As for how the genome changes, we will further conduct whole gene sequencing of the fusant to further analyze the mechanism of the increase in L-lysine production.
References
[1] QU ZH, DING MX, WANG XZ. Cell biology[D]. Beijing: Higher Education Press, 1995. (in Chinese).
[2] WANG WZ. Applied microbiology[D]. Beijing: Light Industry Publishing House, 1996. (in Chinese).
[3] PANG XY, WANG JY. Construction of yeast fusants of directly transform starch into ethanol[J]. Chinese Journal of BIotechnology, 2001, 17(2):165-169. (in Chinese).
[4] GAO NF, WANG SH. Construction of yeast of reducing acid by intergeneric fusion between Saccharomyces bayanus and Schizosaccharomyces pombe[J]. Chinese Journal of Biotechnology, 2000, 16(6): 718-722. (in Chinese).
[5] ZHANG WG, CHEN YF. Breeding of L-lysine producing mutant and studying under fermentation conditions[J]. Biotechnology, 2009, 19(4): 36-40. (in Chinese).
[6] PAEGLE L, RUKLISHA M. Lysine synthesis control in Corynebaterium glutamicum Re115 in mixed substrate (glucose-acetate) medium[J]. J Biotechnol, 2003(104): 123-128.
[7] WITTMANN C, BECKER J. The L-lysinestory: From metabolic pathways to industrial production[J]. MicrobiolMonogr, 2007, 12(5): 39-70.
[8] EGGELING L, SAHM H. L-Glutamate and L-lysine: traditional products with impetuous developments[J]. Appl Microbiol Biotechnol, 1999(52): 146-153.
[9] ZHANG WG. Study on L-lysine fermentation[J]. Fajiao Keji Tongxun, 2005, 34(1) : 7-8. (in Chinese).
[10] ZHANG JL, HU YH. The identification and fermentation conditions' optimizing of a strain L-Lysine producer[D]. Changchun: Jilin Agricultural University, 2008: 3-5. (in Chinese).
[11] WITTMANN C, KIM HM, HEINZLE E. Metabolic network analysis of lysine producing Corynebacterium glutamicum at a miniaturized scale[J]. Biotechnol Bioeng, 2004(87): 1-6.
[12] PFEFFERLE W, MOCKEL B, BATHE B, et al. Biotechnological manufacture of lysine[J]. Adv Biochem Eng Biotechnol, 2003(79): 59-112.
[13] TAKENOUCHI E, YAMAMOTO T, NIKOLOVA DK, et al. Lysine produc-tion by S-(2-aminoethyl) -L-cysteine resistant mutants of Candida pelliculosa[J]. Agric Biol Chem, 1979(43): 727-734.
[14] WEI AY, GE YP. Metabolic pathway of L-lysine to industrial production[J]. Fajiao Keji Tongxun, 2011, 40(2): 31-34. (in Chinese).
[15] HAIDARIS CG, BHATTACHARJEE JK. High lysine excreting mutants of Saccharomyces cerevisiae[J]. J Ferment Technol, 1977, 55(2): 189-192.
[16] TOSAKA O, TAKINAMI K, HIROSE Y. L-lysine production by S-(2-aminoethyl) L-cysteine and α-amino-β-hydroxyvaleric acid resist-ant mutants of Brevibacterium lactofermentum[J]. Agric Biol Chem, 1978(42): 745-752.

- 农业生物技术(英文版)的其它文章
- Report on the Breeding of Dahen 799 Broilers
- Evaluation and Analysis for Survey of Tea Production Quality Safety Management and Control
- Study on the Preparation of "Oil-tea" Instant Tea from the Compound Extract of Green Tea and Ginger
- Research Progress on Chemical Constituents and Pharmacological Effects of Zhuang Medicine Cocculus laurifolius DC.
- Study on Quality Standard of Lujing Yiqi Shengxue Pills
- Research on the Development of Guangxi Zhuang and Yao Ethnic Medicine Industry