
Ⅱ型先天性厚甲症基因变异研究及文献回顾
2022-07-09 08:44卫艳萍李关芳王廷廷张宁宁谢彤阳
河南医学高等专科学校学报 2022年3期
卫艳萍,李关芳,王廷廷,张宁宁,谢彤阳
(焦作市人民医院皮肤科,河南 焦作 454002)
先天性厚甲症(pachyonychia congenital,PC)是一种罕见的常染色体显性遗传性皮肤病。其临床特征为指(趾)甲过度角化肥厚、变黄,可伴有局限性掌跖角化、口腔黏膜白斑、毛囊角化、多发性脂囊瘤、胎生齿及毛发异常等症状。甲损害常于刚出生或出生不久发生,为PC最早的临床症状之一,几乎见于所有患者,主要症状为甲床过度角化、厚甲及甲板弯曲变形[1]。
1 资料与方法
1.1 资料 患儿男,4岁3个月,汉族,双侧手及双足指(趾)甲增厚4 a。患儿出生体质量3.9 kg,足月顺产第一胎,出生时下牙槽即伴有2颗胎生牙,3个月时无明显诱因出现指(趾)甲肥厚,质坚硬,伴年龄增长甲逐渐变黄至污褐色,甲板增厚呈进行性。家族史:家系来自河南省郑州市,父母非近亲结婚,家族中无类似表现患者。体格检查:患儿一般情况可,心肺腹等系统检查未见明显异常。皮肤科检查:患儿双侧手及双足指(趾)甲共18甲(除双手小拇指)显著肥厚呈楔形,黄褐色,质硬不透明,表面粗糙,甲半月存在(图1a、b)。未见局限性掌跖角化,口唇、颊黏膜未见异常,毛发均匀分布,发量稍稀疏。实验室检查:真菌镜检及培养均阴性。常规生化化验未见明显异常。

手指甲损害 脚趾甲损害图1 PC患者的临床表现
1.2 方法
1.2.1 标本采集与DNA提取 本研究获河南省人民医院医学伦理委员会批准[批件号:(2019)伦快审第(07)号],在受试者签署知情同意书后,采集患者及其父母血样。采用TIANamp全血基因组 DNA 提取试剂盒[天根生化科技(北京)有限公司]提取全血基因组DNA。同样方法提取100例无亲缘关系正常人基因组DNA作为对照。
1.2.2 外显子及Sanger测序 使用上海韦翰斯生物医药科技有限公司定制探针(依据OMIM数据库截至2020年5月已知的致病基因),由TWIST合成之后进行全外显子组文库捕获,构建先证者的全外显子组文库。简要操作如下:按照试剂盒操作说明书,配置杂交反应体系,并在PCR仪上进行反应。过夜杂交;按照试剂盒操作说明书,配置捕获反应体系,获得捕获文库;配置扩增反应体系,放置PCR仪上反应。PCR反应程序为:98 ℃ 45 s;98 ℃ 15 s,60 ℃ 30 s,72 ℃ 30 s,循环6次;72 ℃ 5 min,4 ℃ hold;产物经过磁珠纯化后定量,-20 ℃保存。使用PE100模式在MGISEQ-T7测序平台上进行双端测序。对所发现的变异位点应用Sanger测序,在先证者、先证者父母及100名健康对照者中进行验证。所有实验由上海韦翰斯生物医药科技有限公司完成。
2 结果
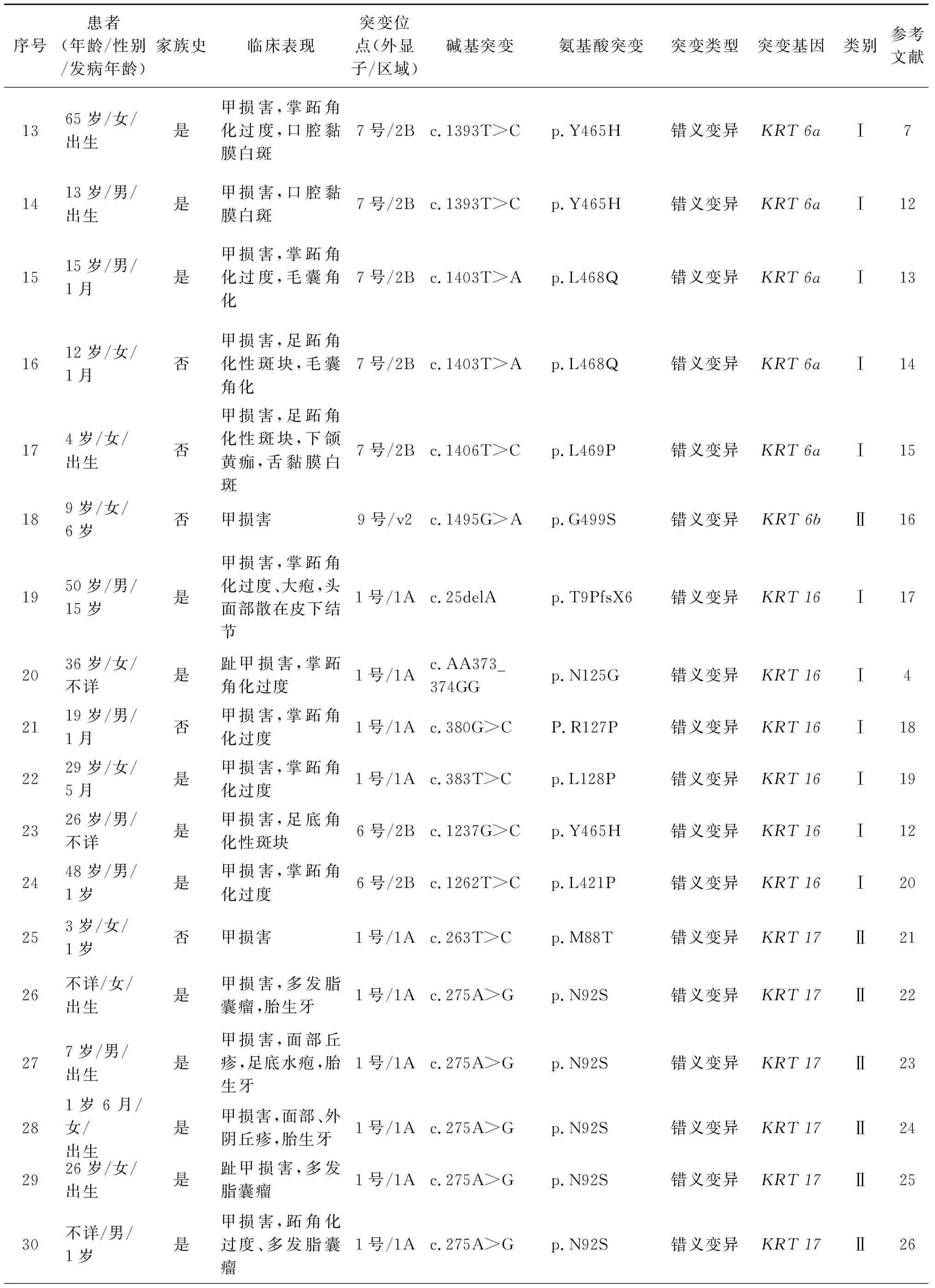
PCKRT基因变异与临床表现,见表1。患者及其父母全外显子测序目标区数据平均测序深度大于100×,超过 99%的目标捕获区域测序深度大于30×,芯片对目标区域覆盖良好。全外显子及Sanger测序发现,PC患者KRT17基因1号外显子存在错义变异c.275A>G(p.N92S),导致KRT17蛋白质第92位天冬酰胺变为丝氨酸,PC患者父母及100名正常人对照组均未发现此变异。见图2a-2c。

表1 PC KRT基因变异与临床表现
续(表1)

续(表1)

图2a PC者KRT 17外显子1第275位碱基DNA序列(A→G箭头处);图2b PC者父亲KRT 17外显子1第275位碱基DNA序列;图2c PC者母亲KRT 17外显子1第275位碱基DNA序列
3 讨论
PC临床上主要分为两型:PC-Ⅰ型和PC-Ⅱ型,PC-Ⅰ型最为常见,又名Jadasshon-Lewandowsky综合征,表现为厚甲、掌跖角化过度、毛囊角化和口腔黏膜白斑,由KRT6a和KRT16突变所致;PC-Ⅱ型又称 Jackson-Lawler 综合征,在甲损害基础上多伴有胎生牙、多汗及多发脂囊瘤,一般无口腔黏膜白斑的表现,由KRT17及其成对表达的KRT6b基因突变所致[2]。根据临床表现及基因检测结果,本研究病例明确诊断为PC-Ⅱ型。
角蛋白是一种结构蛋白,在所有的上皮细胞中表达,不同的上皮细胞及组织表达不同的角蛋白,KRT17和KRT6b蛋白主要表达于甲床、掌跖皮肤、表皮附属器、皮脂腺和毛干等细胞组织中,是构成甲、毛发及表皮的主要细胞骨架成分[6]。 角蛋白结构中有螺旋区和非螺旋区,螺旋区包括1A、1B、2A、2B四个亚区,其中1A区起始端和2B末端在角蛋白二聚体的形成中起着关键的作用[9]。本研究对一个PC患者进行KRT基因变异致病分析,先证者在厚甲的同时伴胎生牙,无黏膜白斑,PCR产物测序结果KRT17基因1号外显子错义变异c.275A>G(p.N92S),导致蛋白质中第 92 位天冬氨酸变成丝氨酸,继而干扰角质形成细胞的主要结构蛋白-角蛋白丝的形成,并影响其稳定性,导致角蛋白空间构象发生改变,影响角质形成,最终引起患者胎生牙、甲增厚等临床表现。该位点位于角蛋白高度保守螺旋亚区1A的起始端,是PC-Ⅱ型较常见的变异位点[27,29]。
本研究回顾了既往国内有关PC基因变异的研究,截至目前共报道PC基因变异研究24个家系和9个散发病例,发现变异位点24个,包括错义变异、缺失变异及剪接位点变异,其中错义变异占比最多83.33%(见表1)。在PC患者中,KRT6a和KRT17基因变异较常见,分别占51.52%、27.27%,这与既往研究基本一致[11]。从表1中可看到,KRT基因1A区均是KRT6a和KRT17基因的变异热点区域,c.275A>G(p.N92S)又明显是KRT17基因的热点变异位点,占比55.56%。这些可能与角蛋白的特殊结构及不同部位的结构功能有关。1A区起始端是角蛋白高度保守区,在角蛋白二聚体的形成中起着关键的作用,该区的变异将阻断角蛋白丝的形成并影响其稳定性[26]。从表1中还可看到PC的临床表型特点,PCⅠ型多有黏膜白斑,PCⅡ型均有较典型多发脂囊瘤表现,这与KRT6a和KRT17两种角蛋白主要表达位置不同有关,所以在临床上可以此鉴别两型PC。
总之,本研究对一个PC-Ⅱ型患者基因检测中发现了KRT17基因1号外显子错义变异c.275A>G(p.N92S),并对PC的基因型与表型做了总结分析,对本病的进一步认识、遗传咨询及优生优育提供了理论依据。
猜你喜欢
纺织报告(2022年5期)2022-11-22
中国畜牧杂志(2022年1期)2022-11-06
电子科技大学学报(2022年5期)2022-10-29
皮革科学与工程(2022年1期)2022-01-15
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
毛纺科技(2021年3期)2021-04-06
中国生殖健康(2020年4期)2021-01-18
东方教育(2017年14期)2017-09-25
