
二次标准加入法校正低氟样品测定偏差的试验研究
2022-07-08 13:57:18陈思涵
煤质技术 2022年3期
陈思涵,隋 艳,杨 妮
(1.煤炭科学技术研究院有限公司 煤炭检测中心,北京 100013;2.国家煤炭质量检验检测中心,北京 100013;3.煤炭资源高效开采与洁净利用国家重点实验室,北京 100013)
0 引 言
氟是重要的环境污染元素之一,在能源、环境、建筑等领域是评估环境生态效应的重要指标[1-7]。氟离子选择电极因其具有灵敏度高、重现性好、干扰少、设备简单等优势,广泛应用于氟离子浓度的测定[8-9],已被列为多种样品的国家标准推荐方法[10-12]。
离子选择电极的工作原理为电极电位与溶液中待测离子浓度的对数呈线性相关关系,即呈能斯特(Nernst)响应,符合能斯特方程。可采用校正曲线法或标准加入法测定样品中氟离子浓度[10-12],对数呈线性相关关系是保证测定结果准确性的先决条件。但当待测液中氟离子浓度较低时,氟离子选择电极的电位响应与浓度变化呈非线性关系,从而造成测定结果发生偏离。在开展固体生物质燃料中氟含量测定方法研究的过程中,发现其含量大多在10 μg/g~30 μg/g,造成待测液中氟浓度大多小于0.5 μg/mL,并影响测定结果的准确度。该现象在测定试剂空白中氟含量的过程中也存在。李中愚等[13]曾提出采用浓度比例尺的方法对低浓度待测样品进行测定,该方法需配置多个不同浓度标准溶液并进行多次测定,且无法消除基体干扰,不适用于煤炭、土壤、固体生物质等基体较为复杂的待测样品;乔正道等[14]曾提出采用等浓度二次标准加入法对低浓度样品进行测定,该方法可将最低定量浓度降低至0.01 μg/mL,但计算过程复杂,不适用于批量样品检测。
笔者以不同浓度氟离子待测液为研究对象,考察了溶液浓度对离子选择电极能斯特响应的影响及对检出限的影响,提出采用不同浓度二次标准加入法校正低浓度氟溶液测定偏差,并分别以低浓度系列氟标准溶液和低浓度基体待测溶液验证方法可行性,以期提高低氟样品测定结果的准确度,为建立以固体生物质燃料为代表的低氟样品中氟含量测定方法提供理论依据。
1 试验部分
1.1 氟离子选择电极
分别选取4个不同型号、不同性能的离子选择电极,试验前均使用低浓度氟溶液进行活化处理。电极A:雷磁PF-1-01型氟离子选择电极,已使用1个月;电极B:雷磁PF-2-01型氟离子选择电极,已使用1年以上;电极C:201型氟离子选择电极,已使用1个月;电极D:201型氟离子选择电极,长时间放置2年以上,未使用。
1.2 待测溶液的制备
1.2.1系列标准溶液
使用预先干燥的优级纯氟化钠,将其配制成浓度为1 000 μg/mL的标准工作溶液,并逐级稀释至0.01 μg/mL~10.00 μg/mL系列浓度梯度的标准溶液。配制过程中,各浓度标准溶液中均加入10 mL 总离子强度缓冲溶液(TISAB,pH=6.0),并定容至100 mL。
1.2.2基体待测溶液
使用煤中氟成分分析标准物质GBW 11121a,其标准值及不确定度为(247±15)μg/g。按GB/T 4633—2014规定的称样量和待测液制备步骤配制100 mL基体待测溶液,基体待测溶液理论浓度为(1.16~1.31)μg/mL。通过吸取不同体积的待测液,配制不同浓度梯度的基体待测溶液。配制过程中,各浓度标准溶液中均加入10 mL 总离子强度缓冲溶液(TISAB,pH=6.0),并定容至100 mL。
1.3 试验方法
1.3.1一次标准加入法测定氟含量
将待测溶液转移至100 mL烧杯中,以氟离子选择电极为指示电极、饱和甘汞电极为参比电极,对待测溶液电极电位进行测定,待电位稳定后记录响应电位值E1,立即加入1.00 mL氟标准溶液,待电位值稳定后记录响应电位值E2。根据GB/T 4633—2014的规定,加入的氟标准溶液浓度应考虑待测液中氟含量(cxVx),以确保加入的标准氟含量(csVs)大于待测液中氟含量的4倍。在试验过程中可根据E1的数值选择加入不同浓度的氟标准溶液,控制ΔE在(20~40)mV。利用电极斜率对待测溶液中氟含量进行计算。
1.3.2二级标准加入法测定氟含量
将待测溶液转移至100 mL烧杯中,以电极最低定量浓度为参考,加入10 μg/mL氟标准溶液5 mL后,以氟离子选择电极为指示电极、饱和甘汞电极为参比电极,对待测溶液电极电位进行测定,待电位稳定后记录响应电位值E′1,立即加入1.00 mL氟标准溶液,待电位值稳定后记录响应电位值E′2。参照GB/T 4633—2014的规定,控制ΔE′在(20~40)mV。利用电极斜率对待测溶液中氟含量进行计算。
2 标准加入法计算原理
离子选择电极的工作原理为电极电位与溶液中待测离子浓度在一定范围内呈能斯特(Nernst)响应,即log(cF)-E呈线性相关关系,见式(1)。
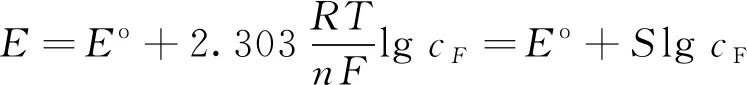
(1)
式中,E为实测电动势,mV;E0为标准电动势,mV;S为电极斜率;cF为待测液中氟浓度,μg/mL。
式(1)中的E0为常数,与待测液基体、电极插入溶液深度等条件有关;在线性范围内S仅与温度、电极表面光洁程度等有关,对于一价离子而言,其理论值为59.2 mV,当其低于55.0 mV时,应更换电极。
当采用一次标准加入法时,待测溶液加标前后电位变化可按式(2)计算。
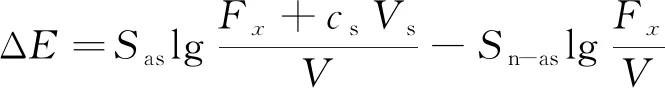
(2)
式中,cs为一次加标浓度,μg/mL;Vs为一次加标体积,mL;ΔE为加标前后电位变化量,mV;Fx为待测液中氟质量,μg;Sn-as为一次加标前待测液中氟离子浓度处的电极斜率;Sas为一次加标后待测液中氟离子浓度处的电极斜率。
当一次加标前后待测液浓度均符合能斯特响应时,Sas=Sn-as=S,式(2)可化简为式(3),即按式(4)直接计算待测液中氟质量Fx。

(3)
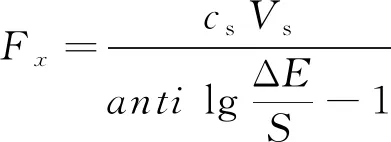
(4)
因此,通常所称的标准加入法,即一次标准加入法均按式(4)简化计算求取待测液氟含量,相关国家标准也均采用该公式进行结果计算。
当采用二次标准加入法时,通过向待测溶液中加入已知浓度的标准溶液,提升待测溶液中氟离子浓度,使电位测定前待测液中浓度可符合能斯特响应。故由式(4)减第一次加标量,即得式(5)计算待测液中氟含量Fx。

(5)
式中,cs,1、cs,2分别为第一次加标浓度和第二次加标浓度,μg/mL;Vs,1、Vs,2分别为第一次加标体积和第二次加标体积,mL;ΔE′为第二次加标前后电位变化量,mV。
3 结果与讨论
3.1 溶液浓度对电极能斯特响应的影响
3.1.1能斯特响应临界浓度
为考察不同电极的能斯特响应情况,明确其能斯特响应临界浓度,分别使用电极A、B、C、D对浓度梯度为0.01 μg/mL~10 μg/mL的标准溶液进行电位测定,电极的响应电位与溶液中氟离子浓度对数的线性回归曲线斜率的绝对值即为电极斜率S。电极在不同浓度范围的电极斜率见表1,不同电极的能斯特响应曲线如图1所示。以相邻5个浓度的线性相关系数r>0.999[15]为能斯特响应临界浓度(cL)以及以斜率S>55.0为符合标准要求的最低定量浓度(cLOQ)分别在图1(a)~(d)中标示。
图1表明不同电极均在低浓度区存在非线性响应区间,能斯特响应临界浓度受电极自身性能状态影响。性能较好的电极能斯特响应临界浓度为0.05 μg/mL~0.07 μg/mL,最低定量浓度均为0.07 μg/mL,与文献中0.05 μg/mL~0.1 μg/mL一致;而长期存放或长期使用后的电极对低浓度氟离子响应不敏感,能斯特响应临界浓度和最低定量浓度均向高浓度方向移动,能斯特响应临界浓度增大至0.10 μg/mL~0.30 μg/mL,最低定量浓度增大至0.30 μg/mL。二者的变化与电极敏感膜氟化镧(LaF3)单晶片的离子交换性能、吸附保留性和溶解度有关,当氟化镧在溶液中溶解度大于待测液浓度时,会造成氟化镧的溶解干扰,从而影响低浓度范围内的电位响应[14]。

图1 氟离子选择电极对不同浓度范围能斯特响应曲线
3.1.2检出限与临界浓度的关系
为考察氟离子选择电极检出限与能斯特响应临界浓度的关系,采用HJ 168-2010等相关标准和有关化学分析-方法检出限等方法[16-18],将校准曲线的直线部分外延的延长线与通过空白电位且平行于浓度轴的直线的交点浓度作为该氟离子选择电极的检出限,以4倍检出限作为该电极测定下限。各电极检出限和测定限见表2。

表2 不同氟离子选择电极检出限与测定下限
结合对表1的分析,由表2的结果分析表明:以电极的检出限和测定下限为指标,将其用于描述电极对低浓度含氟待测液的敏感程度和评估测定结果的准确性时存在一定的局限,无法表述电极老化造成的对低氟溶液不敏感、非线性区间增大的现象。电极的检出限、测定下限均与电极的能斯特响应临界浓度无关。
3.2 测定低浓度标准溶液的偏离及校正
3.2.1测定低浓度标准溶液的偏离
为考察氟离子选择电极采用标准加入法测定低浓度标准溶液时的偏离情况,以浓度梯度标准溶液为研究对象,使用电极D进行氟含量测定试验,以1.00 μg/mL~10.00 μg/mL标准工作溶液绘制电极工作曲线,并以获得的该电极斜率按式(4)对待测标准溶液中氟含量进行计算,即采用一次标准加入法。不同浓度待测标准溶液的测定值与理论值偏离情况如图2所示。

图2 不同浓度标准溶液的测定值与理论值偏离情况
图2表明,当溶液浓度大于0.20 μg/mL时,使用一次标准加入法测定标准溶液中氟含量可确保测定结果的准确度,该浓度与图1中电极D线性响应临界浓度基本一致。而当标准溶液浓度小于0.2 μg/mL时,测定结果显著偏离理论值,氟含量测定值的相对标准偏差达28.4%~364%,且呈现浓度越低则偏差越大的现象。
3.2.2实际斜率计算法对低浓度测定偏离的校正
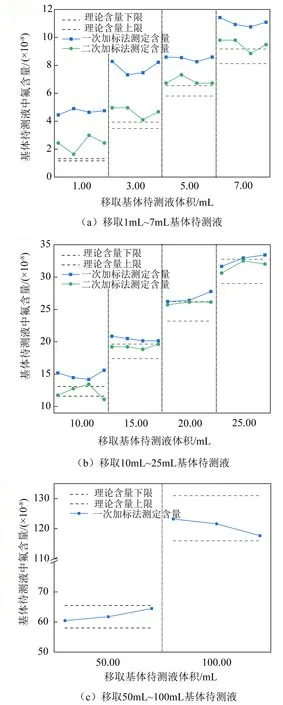
造成低浓度含氟待测标准溶液测值偏差的现象,除电极自身对低浓度待测液不敏感外,与一次标准加入法计算方法有关。由式(2)和图1可知,当待测溶液浓度低于能斯特响应浓度cL时,Sas≠Sn-as,且呈Sn-as 通过查询表1中电极响应曲线确定低浓度处电极响应实际斜率,以待测液浓度数值上处于浓度梯度范围中间段处的斜率为实际斜率,对浓度为0.01 μg/mL~1.00 μg/mL待测标准溶液测定结果按式(2)进行数据处理,结果详见表3,其中加标前、加标后的电极斜率分别用Snb、Sjb表示。 表3数据表明,使用一次标准加入—实际斜率计算法,当浓度大于0.2 μg/mL时,待测溶液中氟含量测定值与理论值偏差较小,与使用式(3)的计算结果一致;当浓度小于0.2 μg/mL时,待测溶液中氟含量测定值的标准偏差由28.4%~364%校正至15.9%~87.7%,在一定程度上校正了氟离子选择电极测定低浓度标准溶液时产生的偏差情况,但仍无法确保测定结果的准确度。 表3 实际斜率法对低浓度待测液氟含量计算结果 3.2.3二次标准加入法对低浓度测定偏离的校正 为解决电极对低浓度待测液不敏感的现象,采用二次标准加入法,即第一次标准液的加入旨在提高待测溶液中氟浓度至能斯特响应浓度范围,第二次标准液的加入旨在读取待测液电位变化以计算待测液中氟含量。采用该方法测定0.01 μg/mL~0.10 μg/mL氟标准溶液,氟含量测定结果见表4,其中测定浓度的数据由100 mL溶液中氟含量计算而得。 表4 二次标准加入法测定低浓度标准溶液中氟含量测定结果 表4数据表明,二次标准加入法的使用可有效校正氟离子选择电极测定低浓度含氟待测液时的偏差,对低浓度标准溶液中氟含量测定的偏差可校正至2.8%~17.8%。当浓度高于0.02 μg/mL时,相对偏差均小于10.0%,大幅提高了低浓度样品测定结果的准确性。 为考察氟离子选择电极采用标准加入法测定低浓度基体待测溶液的偏离情况,以不同稀释倍数基体标准溶液为研究对象,使用电极D分别使用一次标准加入法和二次标准加入法对不同浓度基体待测液中氟含量进行测定,以1.00 μg/mL~10.00 μg/mL标准工作溶液绘制电极工作曲线,并以该电极斜率对溶液中氟含量进行计算。不同浓度基体待测液使用一次标准加入法的测值偏离情况以及使用二次标准加入法的测值校正情况如图3所示。 图3 基体待测液中氟含量测定值与理论值偏离及校正情况 结果表明:使用一次标准加入法测定低浓度基体待测液仍存在测定结果的偏离情况;当基体待测液理论浓度大于0.30 μg/mL时,一次标准加入法测定结果在理论浓度范围内,可保证测定结果对准确度;一次标准加入法准确定量的临界浓度范围与标准溶液测定结果一致。 当使用二次标准加入法时,各浓度级测定结果偏离情况均得到显著改善,可将准确定量的临界浓度拓宽至0.10 μg/mL,即移取10.00 mL即可确保测定结果在理论浓度范围内。但当理论浓度小于0.10 μg/mL时,与标准溶液的校正效果不同,低浓度范围测定结果仍存在高于理论浓度范围的现象。 出现该现象的原因有以下3个方面: (1)由于在测定结果中减去氟加标量,因此在最终结果中存在误差累积的现象,如试剂空白误差、移取体积误差、定容误差、加标误差等,并全部反映在最终结果的偏差情况上,因而造成测定结果偏高并超出理论浓度范围的现象; (2)由于基体效应对电极的灵敏度产生一定的影响,煤高温水解配制的待测液中含有不同浓度的S2-、Cl-等干扰离子,从而影响氟离子测定结果的准确度; (3)由于低浓度理论浓度范围由稀释倍数计算得到,当待测液浓度通过加标提升后,电极自身测定不确定度增大,当电位最小分度值变化0.1 mV时,测定结果即变化0.3 μg,从而造成氟离子测定结果偏差较大。 (1)氟离子选择电极在低浓度区存在响应电位(E)与浓度对数(logC)为非线性关系的现象,能斯特响应临界浓度与电极自身性能有关。长时间使用或放置会造成电极老化,能斯特响应临界浓度和最低定量浓度均向高浓度方向移动,检出限、定量限均无法反映能斯特线性响应极限浓度变化情况。 (2)在非能斯特响应浓度范围内,使用一次标准加入法测定溶液中氟含量存在测值显著偏高的现象,使用实际斜率计算法可改善测值偏离的情况,但效果有限。使用二次标准加入法可显著改善测值偏离情况,将干扰较少的待测液定量测定下限减至0.02 μg/mL,测定相对偏差小于10%;可将干扰较多的基体待测液定量测定下限减小至0.10 μg/mL。 (3)当待测液未加标时的电位响应值低于或近似于能斯特响应临界浓度处的电位响应值时,可采用二次标准加入法提高待测液中氟离子浓度同时消除基体效益的影响。第一次加标量应等于或略大于最低定量氟含量(cLOQVx),避免加标后待测液中氟含量过高,造成电位响应的最小误差不确定度影响测定结果准确度的现象。第二次加标量应控制ΔE′在(20~40)mV。 (4)当氟离子选择电极测定低浓度待测样品时,宜绘制低浓度电位响应曲线图,确定能斯特响应临界浓度和最低定量浓度,分别记录临界浓度、最低定量浓度的电位响应值,并定期进行核查。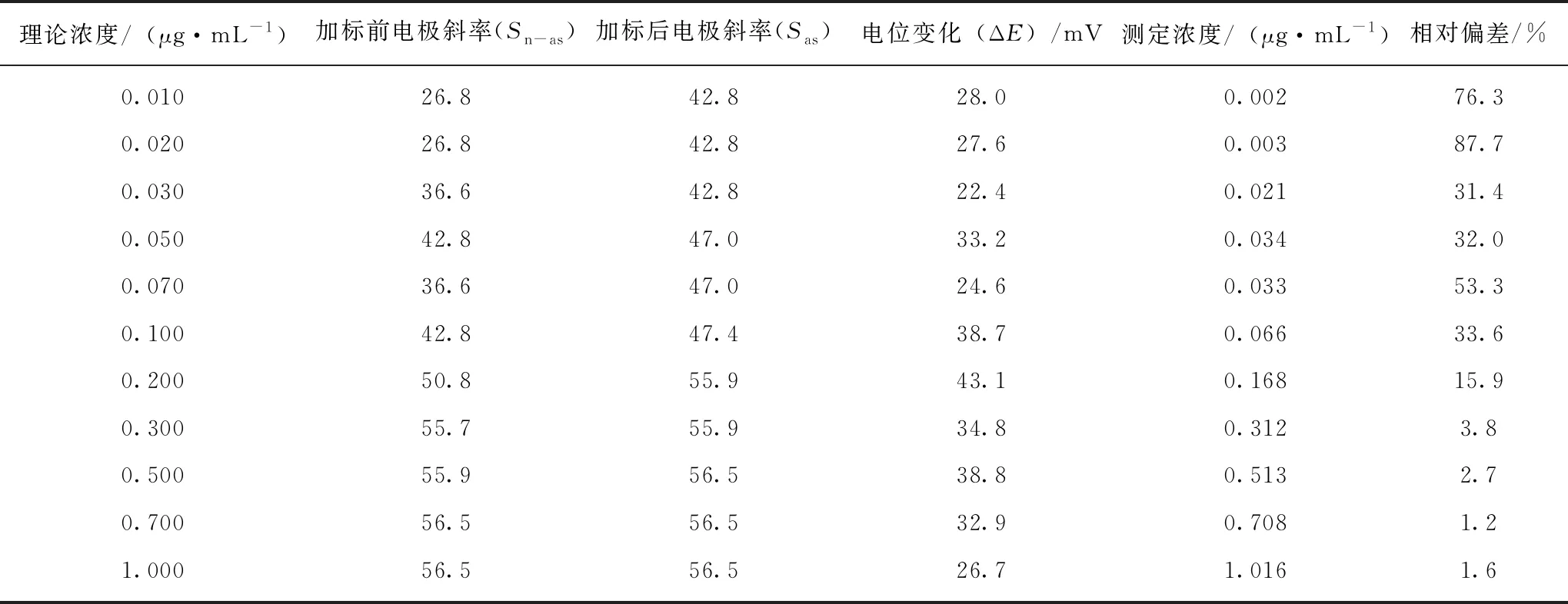

3.3 测定低浓度基体待测液的偏离及校正
4 结 论
猜你喜欢
资源节约与环保(2022年8期)2022-09-20 02:25:58
湿法冶金(2022年2期)2022-03-29 08:44:56
基层中医药(2020年8期)2020-11-16 00:55:14
湿法冶金(2020年5期)2020-10-12 03:00:50
初中生世界·七年级(2017年6期)2017-07-03 19:31:45
初中生世界·八年级(2017年6期)2017-07-03 08:37:59
初中生世界·八年级(2017年3期)2017-03-24 15:49:14
初中生世界·七年级(2017年3期)2017-03-15 20:55:28
无机盐工业(2017年6期)2017-03-11 14:43:47
水利技术监督(2016年6期)2017-01-15 14:01:28
