
177Lu放射性治疗药物研究新进展
2022-06-23 08:45:08杨宇川阚文涛魏洪源卓连刚王关全
同位素 2022年3期
杨宇川,阚文涛,杨 夏,魏洪源,卓连刚,王 静,廖 伟,赵 鹏,王关全
(1.中国工程物理研究院 核物理与化学研究所,四川 绵阳 621999;2.国家卫生健康委员会核技术医学转化重点实验室(绵阳市中心医院),四川 绵阳 621099;3.核医学与分子影像四川省重点实验室,四川 绵阳 621999)
作为医用核素,177Lu因其本身的诸多特性而引起放药领域的广泛关注,如其衰变产生低能β-粒子,半衰期为6.646 d。在制备方面,多个研究型反应堆可以制备得到高比活度177Lu,易得性也使得其作为靶向治疗核素引起广泛关注。与此同时,177Lu衰变过程中产生的伽马射线及其能量特性使其适合应用于单光子计算机断层扫描(SPECT),所获得的图像可用于获取放射性药物的靶向性、治疗前的成像与肿瘤及组织器官吸收剂量的评估、药物代谢动力学以及排泄行为,因此177Lu也可以作为诊疗一体化核素应用。本文综述了近两年177Lu作为治疗核素及诊疗一体化核素的临床前研究及临床应用的最新研究进展。
1 177Lu放射性药物的标记技术
177Lu作为镧系金属元素,其衰变示意图示于图1。在标记方面,pH和温度是重要参数,配体与Lu3+配位的路易斯碱的质子化作用会极大程度的削弱配位率,因此通常在较高pH条件下的配位反应会较快。但是在pH>6时,Lu3+会形成稳定的氢氧化物,为确保没有沉淀产生,获得较高的标记率,Lu的标记反应在pH为4~6区间进行[1]。在标记结束后,标记物在pH>6的环境中稳定存在[2]。对于DOTA,在25~37 ℃条件下标记需要1 h以上,在80 ℃以上反应,标记时间需要10 min以上。对于其他配体如CHXA-DTPA及1B4M-DTPA,通常在4~25 ℃条件下标记需要进行1 h。总的来说,较低的温度及较短的标记时间可以避免非必要的降解,尤其对热敏感的蛋白尤为重要。
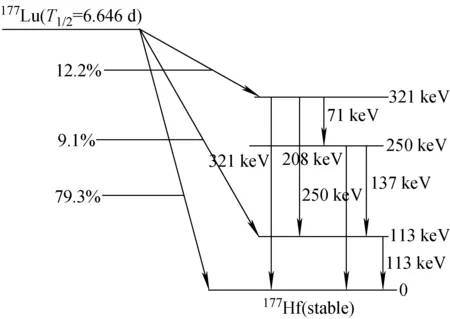
图1 177Lu衰变示意图Fig.1 The scheme of 177Lu decay
Lu与几种常见配体在25 ℃,0.1 mol/L HNO3条件下形成的金属配合物的结合常数lgKb分别为:177Lu-EDTA为19.65,177Lu-DTPA为22.44,Lu-DOTA为25.4。177Lu-TETA在25 ℃,0.2 mol/L NaNO3条件下形成的金属配合物的结合常数lgKb为15.31。相比之下,DOTA与177Lu形成的配合物的热力学稳定性优于其他常用的脂肪类或环状氨基羧酸酯类配体与177Lu形成的配合物[3]。虽然稳定常数很高,Lu3+与DOTA及其他大环配体的配位反应速度很慢,特别是在177Lu放射性药物制备时,177Lu及配体的浓度一般都很低。这是177Lu标记药物在制备方面的一大劣势。对于多肽类药物,为获得较高的标记率,需将177Lu的标记温度提升到95 ℃以上,可以将标记时间缩短至25~30 min,如果在25~37 ℃条件下,则要反应1 h以上。
2020年,Srensen等采用Trasis miniAllinone完成了符合GMP标准的177Lu-PSMA自动化合成,可与68Ga-PSMA协同完成对前列腺癌的诊疗一体化。标记反应在2 mL pH为4.5抗坏血酸溶液中进行,120 ℃反应100 s,105 ℃反应800 s,制备过程总历时30 min,放化纯度大于98%,校正后的最高活度为15.3 GBq[4]。Eryilmaz等在2021年采用Modular Lab Easy (ML Easy)自动化合成177Lu标记的纤维活化蛋白特异性抑制剂FAPI。在pH为4.5的抗坏血酸缓冲体系中,在95 ℃反应20 min即可完成,产物177Lu-DOTA-FAPI-04标记率为(88±3)%;177Lu-DOTA-FAPI-46标记率为(86±3)%,放化纯度大于99%,标记物可稳定保持4 d[5]。FAPI可用于乳腺癌,卵巢癌等多种肿瘤的治疗,首次完成可重复的177Lu-FAPI的自动化合成为该药的制备提供了便利。
考虑到抗体对高温的耐受力有限,177Lu标记DOTA修饰的抗体时,标记温度只能够控制在60 ℃来缩短标记时间,同时获取高标记率。Sinnes等[6]在2019年考察了新配体AAZTA与不同三价金属的配位效率,如图2所示,AAZTA是一个混合配体,兼具环状与芳香族配体的特性。不同于大环配体DOTA,AAZTA与177Lu形成标记物的几何构型与DOTA不同。对于DOTA,用于配位的原子从N2O2到N2O4不等,其中的一个羧基集团用于靶向分子的双功能化,对于三价放射性金属,DOTA与其形成的复合物整体呈现电中性。而AAZTA用N3O4核与Lu形成整体静电荷为-1的配合物。整体的负电荷是由于四个去保护的羧基用于与放射性金属Lu进行配位,四个羧酸集团的负电荷过度代偿了放射性金属Lu的3价正电。AAZTA在温和条件下可以标记三价的放射性金属如68Ga,44Sc及177Lu。Sinnes等评价了多肽TOC与配体AAZTA形成的复合物AAZTA-TOC与68Ga,44Sc及177Lu的配位能力与稳定性[6]。AAZTA-TOC在温和条件下可以短时间内完成177Lu的放射性标记,制备得到177Lu-AAZTA-PSMA-617,室温下标记率大于99%,标记时间小于5 min,在人血清、PBS、EDTA、DTPA溶液中1 d后仍然稳定。177Lu-AAZTA-PSMA-617对PSMA阳性的LNCap细胞的抑制常数Ki为8~31 nmol/L,与PSMA相当。在内化比例方面,177Lu-AAZTA-PSMA-617对GCP-1的细胞内化率为13%~20% IA/106细胞,PSMA-617对GCP-1的细胞内化率为17%~20% IA/106细胞[7]。从标记温度、标记时间、标记率、标记物稳定性以及细胞层面的生物性质来看,新配体AAZTA合格,替换DOTA后,在体外对LNCaP细胞的亲和性未呈现负面影响,同时还提供了快速而温和的标记条件,具备进入自动化合成研究的条件,可以使AAZTA-PSMA-617应用到活体的诊疗一体化当中。但若考虑在临床上以AAZTA替代DOTA,除考虑AAZTA的放射性标记性质,177Lu-AAZTA-PSMA-617的体外稳定性及体外靶向前列腺癌的特性外,还应评价其在活体中对前列腺癌的靶向能力和代谢情况。

图2 AAZTA-PSMA-617结构示意图Fig.2 Molecular structure of AAZTA-PSMA-617
2 临床前177Lu放射性药物
2.1 177Lu标记的纳米颗粒
近几年放射性标记纳米颗粒迅速发展成为微肿瘤领域研究的热点。纳米科技、放射化学及同位素生产推动了放射性标记的纳米颗粒在肿瘤诊断及治疗方面的应用。纳米颗粒的尺寸设计符合肿瘤的脉管系统,确保在活体用药时非靶点部位泄露的药物量最小化。相比于其他种类的放射性药物,放射性标记的纳米颗粒的优点在于其可以提供快速、高效及放射化学稳定的放射性标记而不改变纳米颗粒固有的药代动力学特性[8-10]。在纳米颗粒表面修饰多功能的靶向基团或多功能的治疗性基团可以实现纳米颗粒的功能化。在众多纳米颗粒当中,金纳米颗粒在近红外区有吸收,当其处于激光辐照或射频场中时能够对细胞造成不可逆的热损伤[11]。177Lu标记纳米颗粒是多功能化的靶向放射性药物,同时充当着放疗、热消融及多方式成像试剂的角色。
2021年,Kogos等以177Lu标记血栓调节蛋白抗体(mAb-201b)偶联的金纳米颗粒用于诊疗一体化研究,通过小鼠模型SPECT显像验证抗体偶联金纳米颗粒放射性药物的肿瘤靶向性。在纳米颗粒上偶联抗体,在鼠模型内实现了特定抗原血栓调节蛋白的靶向,未对周围正常组织造成影响[12]。临床研究的经验表明,相比多个靶向基团修饰的纳米颗粒,单个靶向基团修饰的放射性药物的靶向性会有所减弱,同时肿瘤细胞还可能表现出对放射性药物的抗性。2020年Yook等采用177Lu标记的双靶向纳米药物增强金纳米颗粒的靶向性,提高对乳腺癌的治疗效果。Yook等用曲妥单抗及帕尼单抗修饰直径为15 nm金纳米颗粒,实现对HER-2以及EGFR的双靶向;同时对金纳米颗粒进行PEG3000修饰后连接DOTA,进行177Lu标记。在双靶向的放射性纳米颗粒作用下HER-2以及EGFR双阳性的MDA-MB-231-H2N细胞存活率仅为(18.8±1.0)%,而单靶向的放射性纳米颗粒作用下细胞存活率均大于50%。对仅HER-2阳性的BT-474以及仅EGFR阳性的MDA-MB-468细胞,双靶向放射性纳米颗粒的细胞毒性均大于单靶向放射性纳米颗粒。同时,乳腺癌细胞核展现了36~119 Gy的高吸收剂量。该项研究有望解决对HER-2靶向放射性药物有抗性的乳腺癌的治疗问题[13]。但由于采用重金属,在临床应用时的生物适应性仍需要在该种药物进入临床阶段时进行考察。
考虑到采用重金属的生物相容性问题,放射性标记纳米材料可以采用如硅基及仿生材料等生物相容性高的材料。Zhang等在2020年采用177Lu标记的α-促黑素细胞激素功能化的超小硅壳核纳米颗粒用于黑色素瘤的治疗。硅壳核纳米颗粒以PEG及荧光素修饰,再将每个纳米颗粒的表面修饰10~15个α-促黑素细胞激素(αMSH)环状肽类似物。177Lu-αMSH作为整个纳米颗粒的靶向功能基团靶向恶性肿瘤的黑素-1受体[14],其在核素靶向治疗方面的有效性已经在临床前研究中得到了验证[15]。放射性标记纳米颗粒对MCI-R(+)细胞产生次纳摩尔亲和性及高效的内化作用。在Mα1肿瘤小鼠模型的体内分布方面,肿瘤的摄取率在注射后4 h为(5.47±2.5)%ID/g,24 h为(9.29±4.74)%ID/g。在B16F10小鼠模型中,4 h和24 h后的肿瘤摄取率分别为(10.4±3.56)%ID/g和(9.56±1.57)%ID/g。Mα1肿瘤小鼠模型在给药4~24 h后,肿瘤部位的放射性摄取持续上升,在肿瘤部位的滞留时间可长达96 h。放射性标记纳米颗粒经肾脏排泄,96 h后可将80%以上的放射性剂量排出体外。接受0.5 mCi该标记纳米颗粒治疗的B16F10肿瘤小鼠存活时间延长至25 d,并提高了存活率,在18 d时存活率仍为100%[16]。Chakravarty等在2020年仿生合成了内含177Lu的混合微球用于黑色素瘤B16F10的治疗,采用人血清蛋白介导的生物矿化过程,内在标记[177Lu]Lu2O3纳米颗粒于蛋白框架之内。[177Lu]Lu2O3-HSA对黑色素瘤B16F10细胞具有特异性亲和力,体内分布研究显示肿瘤在4 h的摄取率为(11.7±2.1)%ID/g,在肿瘤中滞留7 d,体内其他组织清除快,在治疗效果方面可抑制肿瘤达21 d[17]。
2.2 177Lu标记的多肽
177Lu放射标记多肽靶向在肿瘤表面特异性表达的受体。考虑到靶向血管内皮生长因子(VEGF)的177Lu标记单抗[18]及抗体的单链重组衍生物[19]在鼠模型中展现了肿瘤抑制作用,新近的研究致力于探索具有靶向VEGF新机制的177Lu标记的多肽。神经磷脂-1 (NRP-1)是存在于多种类型癌细胞的表面受体,其过表达及其与血管内皮生长因子-165 (VEGF165)的相互作用与肿瘤的生长和转移密切相关。因此,阻断VEGF165/NRP-1的相互作用是对NRP-1相关病理进行显像和治疗一种很有前景的策略。在2022年,Masowska等设计并合成了两种已知的VEGF165/NRP-1复合物的多肽抑制剂A7R及其较短的类似物枝状肽类Lys(hArg)-Dab-Pro-Arg,并以68Ga和177Lu放射性核素标记分别用于诊断和治疗。在模拟人体体液中标记物的稳定性良好,然而由于酶降解作用导致其在人血清中稳定性不足,需要对肽结构进行修饰提高其稳定性[20]。在研究放射性标记的受体拮抗剂多肽方面,采用合理的多肽结构能够有效避免酶降解作用,从而提高其体内稳定性,如Behnammanesh等在2020年采用177Lu标记生长激素抑制剂受体拮抗剂DOTA-p-CI-phe-cyclo-cn-Cys-L-BzThi-D-Aph-Lys-Thr-Cys-D-Tyr-NH2用于神经内分泌瘤的治疗。生长激素抑制素由于内生多肽酶的作用,血浆半衰期仅为3 min,研究中采用合成的多肽在避免多肽酶降解的同时,保留了多肽的特异靶向性。标记多肽具有高亲和性,Kd=12.06 nmol/L,Bmax=0.2 pmol/106细胞,内化率小于5%。肿瘤模型中4 h后有效的肿瘤摄取率为7.3%ID/g,血液清除较快,靶/非靶比高[21]。该标记多肽具有良好的体内生物学性质,但作为治疗药物对肿瘤的治疗抑制作用的评价需要做进一步补充。放射性标记的环状RGD多肽及其多聚体在肿瘤显像方面的应用较多,但由于治疗性药物要求具有肿瘤高摄取、长滞留时间以及对非靶组织的低放射性浓聚等特性,制约着RGD在肿瘤治疗方面的应用,尤其是较高的肾脏摄取。2013年,鼠肿瘤模型中验证了177Lu-DOTA-E(cRGDfk)2对肿瘤的特异性富集后[22],2020年Pirooznia等不仅对多肽的体内生物学性质进行了评价,还对小细胞肺癌的治疗效果进行了评价。在靶向性方面,靶向肿瘤细胞的特异性结合大于95%。肿瘤鼠模型体内分布显示,静脉注射放射性药物4 h后,肿瘤中达到最大富集,肾清除快。治疗实验中,鼠模型的肿瘤体积发生明显缩小[23]。同时,177Lu-DOTA-E(cRGDfk)2与68Ga-DOTA-E(cRGDfk)2组合进行的放射性诊疗一体化也已经在临床上得到验证,丰富了RGD多肽在临床诊疗一体化的应用[24]。鉴于趋化因子-4受体(CXCR-4)在肿瘤的生长及转移中发挥了重要作用,早期研究采用68Ga标记靶向配体用于CXCR-4阳性肿瘤的显像诊断[25],后拓展到177Lu及90Y用于CXCR-4阳性肿瘤的多肽靶向治疗[26],在采用18F-FDG验证了临床上177Lu及90Y用于CXCR-4多肽靶向治疗的效果后,该策略新型放射性药物的临床前研发也陆续跟进。ávila-Sánchez等在2020年制备了177Lu-CXCR4-L,考察了其对表达趋化因子-4受体(CXCR-4)的乳腺癌及恶性胶质瘤的治疗,采用CXCR-4阳性的Du-4475乳腺癌细胞及C6恶性胶质瘤的鼠模型进行体内性质评价。标记物的Kd<10 nmol/L。鼠模型的体内分布显示,注药96 h后,Du-4475乳腺癌细胞及C6恶性胶质瘤鼠模型肿瘤部位的放射性摄取率分别为(1.95±0.48)%ID/g,(1.57±0.17)%ID/g,此时其他器官的放射性几乎完全排出[27]。在Du-4475乳腺癌细胞及C6恶性胶质瘤鼠模型中,几乎完全排出,肠及肾的放射性摄取接近或略高于肿瘤组织,直到48 h才有所下降。放射性标记多肽的结构会对其生物性质产生影响,其中包括多肽本身、连接剂及配体与放射性核素形成的配合物。2020年,Ding等研究了小细胞肺癌细胞对不同侧链长度的177Lu标记环状NGR的摄取量,虽然不同结构的标记多肽的A549细胞摄取均可观,但由于NGR多肽在转化中会形成isoDGR,其含量将改变肿瘤细胞对多肽的摄取特性,因此可通过改变NGR的侧链长度调节小细胞肺癌细胞对标记多肽的摄取[28]。但该研究仅停留在细胞研究水平,在活体中由于代谢等其他因素,侧链长度是否仍然能够影响多肽的肿瘤摄取,还需要进一步验证。
在SSTR受体放射性多肽靶向治疗方面,最初90Y-DOTATOC及90Y-DOTATATE因为治疗效果理想得以普遍应用。为提高治疗效果,研究拓展β核素177Lu,177Lu-DOTATATE受到关注,而DOTATOC仅用来制备68Ga-DOTATOC与177Lu-DOTATATE组合进行诊疗一体化研究[29]。Krebs等在2020年采用正位嫁接神经内分泌瘤鼠模型,比较两个用药循环的SSTR拮抗剂177Lu-DOTA-JR11以及SSTR激动剂177Lu-DOTATOC的治疗效果。结果显示,在辐射敏感期G2/M期的细胞累积及肿瘤体积缩小方面,拮抗剂均强于激动剂;鼠模型存活时间上,拮抗剂为207 d,激动剂为126 d;肿瘤放射性摄取方面,拮抗剂是激动剂的4倍;在第一个用药循环,拮抗剂的肿瘤/肾放射性摄取比是激动剂的3倍;第二个用药循环上,激动剂的肿瘤摄取率降低而拮抗剂保持稳定[30]。总体来看,拮抗剂的效果均优于激动剂。
177Lu-DOTATATE的NETTER-1临床实验[31]直接促进其得到欧洲药监局及美国药监局的批准。为进一步加强177Lu-DOTATATE的临床治疗效果,近两年其与化疗药物进行联合治疗的协同方案得以开发,采用化疗药物作用于肿瘤细胞核的DNA,协同放射性药物的射线作用杀死癌细胞。2020年,Cullinane等采用177Lu-DOTA-TATE联合小分子聚(ADP-核苷)聚合酶-1他拉唑帕尼抑制剂对二者的联合治疗效果进行临床前评价。在表达SSTR2的AR42J细胞模型中,联合用药方式比单独使用177Lu-DOTA-TATE产生更多的DNA双链断裂,进一步采用AR42J活体鼠模型显示联合用药的抗癌效力明显强于单独使用177Lu-DOTA-TATE[32]。
GRPR拮抗剂相比于相应的激动剂对肿瘤细胞的特异性亲和力高,副作用小,可以弥补其对肿瘤细胞的摄取不及激动剂的劣势。所以GRPR拮抗剂常作为对该受体放射性多肽靶向治疗的首选,有报道筛选生物性质更佳的GRPR受体拮抗剂进行核素多肽靶向治疗。例如,2017年GRPR拮抗剂NeoBoMB1由于其对GRP受体具有纳摩尔范围的亲和性及高活体稳定性得到开发,177Lu-NeoBoMB1及68Ga-NeoBoMB1的肿瘤摄取都很高,且对肿瘤的显像也很清晰。68Ga-NeoBoMB1的临床显像效果好,即将开展177Lu-NeoBoMB1治疗效果的临床评价[33-34]。在这个背景下,2020年Rousseau等比较了GRPR拮抗剂177Lu-ProBoMB1以及177Lu-NeoBoMB1作为表达GRPR前列腺癌治疗药物的生物性质。在细胞层面,177Lu-NeoBoMB1对肿瘤细胞GRPR受体特异性结合(Ki=(2.26±0.24) nmol/L)优于177Lu-ProBoMB1(Ki=(30.2±3.23) nmol/L)。177Lu-ProBoMB1在PC-3鼠模型中,注射后1、4和24 h、肿瘤摄取率分别为(3.38±1.00)%ID/g、(1.32±0.24)%ID/g和(0.31±0.04)%ID/g,均小于相同时间点的177Lu-NeoBoMB1的肿瘤摄取,同时在肿瘤与肾脏的放射性摄取比方面,177Lu-NeoBoMB1的摄取比高。总体来看177Lu-NeoBoMB1比177Lu-ProBoMB1的治疗效果好[35]。尽管在二者比较中,总体治疗指数高的177Lu-NeoBoMB1被筛选出来,但其高卵巢摄取是一大缺陷。如何通过改变连接剂或者改变核素克服卵巢高放射性吸收将成为其进一步开发所面临的一项挑战。
2.3 177Lu标记的抗体
在放射免疫治疗中,与抗体搭配的核素应考虑其半衰期及发射粒子的能量。通常,半衰期为几天的核素由于跟抗体的生物摄取时间匹配,可用于放射性标记抗体的免疫治疗。与90Y(半衰期为2.7 d)相比,177Lu(半衰期为6.647 d)更有利于统筹放射性标记抗体的制备及制定治疗计划。同时,由于177Lu的β-粒子的能量较低,造成的骨髓毒性较低,对周围正常细胞的辐射剂量低,肾毒性也较低。
Yeh等2020年采用177Lu标记磷脂酰肌醇聚糖-1抗体(177Lu-Miltuximab),在Du-145鼠模型体内观察其对前列腺癌的免疫治疗。鼠模型单剂注射6 MBq的177Lu-DOTA-Miltuximab,7 d后,肿瘤部位的浓聚(6.5 ID/g)大于其他正常组织。单剂注射放射性药物比其非放对照产生了更佳的肿瘤抑制效果,单剂注射10 MBq的177Lu-DOTA-Miltuximab可将小鼠的存活天数从45 d延长至85 d[36]。标记抗体具有放射免疫治疗效果,但需较长时间方可观察到肿瘤的放射性浓聚。为保留良好的靶向性并缩短形成明显肿瘤信号所需要的时间,Delage等2020年采用肿瘤上皮标记物-1(TEM-1)scFv-Fc融合抗体1C1m-Fc,与p-SCN-Bn-DOTA偶联后标记177Lu,标记后的抗体免疫反应性82%~90%。体内分布实验显示,标记抗体在(TEM-1)+肿瘤细胞中有特异性摄取,且摄取量与每个抗体上DOTA数量正相关。在SPECT显像中,注药72 h后(TEM-1)+的肿瘤部位的信号比(TEM-1)-的肿瘤部位的信号强1.9倍[37]。通过调节每个抗体上的配体DOTA数量可以增强(TEM-1)+肿瘤部位的放射性摄取,但标记抗体从进入体内到在靶向部位形成对比信号的时间较长,还应该改进抗体的药代动力学性质。
在免疫治疗当中,采用放射性标记的大分子抗体时,达到理想的治疗效果和较长血液循环造成的血液毒性是两个对立的因素[38],可以选择小分子抗体或者抗体片段来打破这一局限[39]。较小的分子尺寸使得其快速的血液清除,并且快速均匀的实现在肿瘤组织中的分布,可以较早的实现肿瘤组织接受较高的辐射,减轻血液毒性,考虑核素的衰变,以上特性均有利于放射免疫治疗发挥其优势。
2020年Tsai等采用177Lu标记PSCA微抗体A11制备177Lu-DTPA-A11,其对22Rv1-PSCA肿瘤小鼠模型进行放射免疫治疗,试图用微抗体代替抗体,实现在肿瘤部位的快速富集,评价其作为表达前列腺干细胞抗原(PSCA)的前列腺癌治疗药物的效果。静脉注射放射性药物8 h后,肿瘤的放射性摄取值达到最高,但注射后72 h,肝肾的放射性仍较高。在细胞毒性方面,注射0.19 MBq的放射性药物168 h后,22Rv1-PSCA肿瘤细胞的存活率仅为24%。作为对照, 虽然I-131标记的微抗体及非放射性微抗体A11的治疗效果比177Lu-DTPA-A11[40]抑瘤效果好,但肾清除较慢,其代谢性能会成为临床应用障碍。其他研究中,采用抗体片段试图改善药代动力学,但仍未能解决肾脏清除慢的问题。Carpanese等2020年为解决177Lu-PSMA抑制剂在药代动力学及计量学方面的缺陷,采用177Lu标记DOTA修饰的lgGD2B抗体的单链变异片段(177Lu-ScFvD2B)对表达PSMA的前列腺癌进行免疫诊疗。177Lu-ScFvD2B对PSMA阳性的LNCap的亲和性及内化效果均优于PSMA阴性的PC-3细胞。在健康裸鼠中,药代动力学性质显著改善,非靶组织清除快,肝部的累积少,但肾的清除较慢。以肺微小肿瘤小鼠模型进行的micro-SPECT/CT显示,在注射初期,LNcap肿瘤组织的摄取率较低,随后逐步提高,在192 h达到最大值。虽然肾清除慢的问题仍然存在,但该放射性药物展现了肿瘤摄取率高、肿瘤滞留时间长的优势[41]。Borgna等为了解决肾清除慢的问题,2020年采用清除速度快的小分子PSMA抑制剂(PSMA-11, 2-PSMA, ZJ-43)减少白蛋白结合的放射性药物177Lu-PSMA-ACB-56高肾摄取。采利用PSMA-11时,表达PSMA的前列腺癌鼠模型中的肾摄取率从(35%~57%) ID/g降低到了(8.3%~9.9%) ID/g。用2.5倍过量的2-PSMA会提高肿瘤/肾脏的摄取比值[42]。小分子抗体片段及微抗体经肾脏排泄,由于放射性的代谢产物在肾细胞中的累积,造成的肾脏毒性是其应用中的劣势。有研究采用了与注射177Lu标记的微抗体同时注射肾保护剂的方式降低了肾脏的放射性累积,在治疗后未发现有肾脏的炎症、细胞凋亡以及坏死[43]。
2.4 177Lu标记的其他分子
临床上对三阴乳腺癌(TNBC)的治疗缺乏有效方案,Hernandez等2020年采用177Lu标记肿瘤靶向分子烷基磷酸胆碱(NM600),实现对TNBC有效的放射性核素靶向治疗。利用同源性4T07(非转移性)/4T1(转移性)肿瘤鼠模型,给药后,4T07(非转移性)和4T1(转移性)肿瘤的剂量分别为(2.04±0.32) Gy/MBq和(1.68±0.06) Gy/MBq,均大于肝的(1.28±0.09) Gy/MBq以及骨髓的(0.31±0.05) Gy/MBq。注射治疗剂量后,鼠模型耐受程度高,177Lu-NM600显著抑制了鼠模型的肿瘤生长并延长了存活期[44]。该药物在两种鼠模型中,表现出优秀的肿瘤靶向性及快速的正常组织清除,安全性高,未出现副反应,为推进临床应用打下了良好的基础。
对骨具有高度亲和力的双磷酸化合物最早被99mTc标记后用于骨显像,双磷酸化合物兼具靶向基团和配体的角色[45]。近年来,核素的应用从99mTc拓展到了177Lu,如177Lu-BPAPD, Lu-177-BPAMD[46]及177Lu-DOTA-zol[47]。Zakaly等2020年通过理论计算评价了177Lu-MDP和177Lu-EDTMP用于骨转移的疼痛缓解治疗的可能性,计算得出177Lu-MDP和177Lu-EDTMP的骨剂量分布高于其他器官,并且放射性标记物能够快速浓聚在骨骼[48]。但这两种磷酸配体的177Lu放射性标记物在活体模型的生物学分布均需要实验评价。99mTc-MDP、153Sm-EDTMP已分别用于骨显像和骨转移的治疗,二者的体内分布具有相似性,均快速的累积在骨组织中,注射6 h后,50%的放射性从血液清除由肾脏排出[49-51]。二者可以搭配应用于骨转移造成的疼痛缓解的诊疗一体化。如果应用177Lu,有望实现单一核素的骨疼痛缓解诊疗一体化,同时避免153Sm-EDTMP使用过程中出现的“饱和效应”而引起的放射性核素153Sm在肝脏的过多累积[52]。
3 临床研究
3.1 177Lu标记的抗体
177Lu-J591作为放射性免疫治疗的药物,其中人源化单克隆抗体J591能靶向PSMA,将177Lu特异性输送到前列腺肿瘤部位,实现对恶性前列腺肿瘤的靶向辐照,很快就进入了临床研究阶段[53]。目前177Lu-J591正处于前列腺癌治疗的临床ⅡB研究中,有望开发成为靶向放射性治疗药物,以在肿瘤发展早期根除微转移病灶。目前的临床研究有望确定177Lu-J591在转移性去势抵抗性前列腺癌(mCRPC)微转移阶段对生存期的影响[54]。
在治疗转移性去势抵抗性前列腺癌方面,除小分子177Lu-PSMA靶向配体药物受到临床关注外,177Lu-J591在临床研究中显著减少了前列腺特异性抗原[55],但其单独用药的效果与小分子177Lu-PSMA靶向配体药物相比略有逊色[56]。鉴于此,Batra等2020年对mCRPC病例进行多西他赛联合177Lu-J591治疗的临床1期试验,考察安全性、剂量极限性,以及最大承受剂量。临床研究的结果显示,分次使用177Lu-J591联合标准剂量的多西他赛的用药方案可行,可以实现精准靶向,初步显现出有效的治疗效果[57]。
3.2 177Lu标记的多肽
近两年,177Lu多肽靶向治疗的临床研究针对多种肿瘤病例,研究的内容为临床用药剂量的探索。在临床前研究中发现靶向成纤维细胞活化蛋白(FAP)的FAP-2286在肿瘤中的吸收高,滞留时间长,且背景活性低。在2020年,Baum等首次在人体中使用了177Lu标记的 FAP-2286用于多肽靶向放射性治疗,评估了其生物分布和动力学,并获得了首次剂量学数据。10例晚期腺癌患者耐受良好,其中3例患者出现自限性头痛,未出现严重短期副作用。患者全身平均吸收剂量为0.05~0.1 Gy/GBq,其中红骨髓平均吸收剂量为0.04~0.09 Gy/GBq,肾平均吸收剂量为0.6~0.9 Gy/GBq,与177Lu DOTATOC的多肽靶向放射性治疗相当。所有患者在延迟显像(长达10 d)上表现出高特异性肿瘤摄取和长时间滞留。该研究证明了177Lu标记的 FAP-2286靶向治疗的可行性。由于肿瘤保留时间较长,该治疗策略是一种非常有前途的广谱癌症治疗选择[58]。在2021年,Ballal等首次采用肿瘤病例评价了177Lu-DOTA.SA.FAPi 以及177Lu-DOTAGA.(SA.FAPi)2在人体中的生物学分布,药代动力学以及计量学。相比于单体177Lu-DOTA.SA.FAPi,同型二聚体177Lu-DOTAGA.(SA.FAPi)2的肿瘤滞留时间显著延长,在结肠和肾脏的摄取与单体相比较高,但耐受性良好。同型二聚体177Lu-DOTAGA.(SA.FAPi)2因其优良的特质,如内化速度快,肿瘤亲和力强,肿瘤组织中保留时间长以及从非靶组织清除快,给各类晚期癌症患者提供了新的治疗方案[59]。GRP多肽在上世纪70年代早期就被发现,177Lu直到30年后方才应用到GRP多肽的标记,并进行了临床研究,如177Lu-AMBA。在此之后,177Lu标记的GRPR激动剂以及拮抗剂陆续进入了临床研究[60]。Kurth等2020年评价了胃泌素释放多肽受体拮抗剂177Lu-RM2在GRPR前列腺癌治疗中的人体剂量学。177Lu-RM2平均给药剂量为(4±0.9) GBq,在给药第1 h后,肿瘤病变部位及胰腺的摄入量有增加,肿瘤的平均吸收剂量为(6.2±3.00) Gy/GBq,胰腺为(1.08±0.02) Gy/GBq,其他器官如肝、肾胆、囊壁、脾以及红骨髓均小于1 Gy/GBq。研究结果显示,177Lu-RM2肿瘤摄取高,血清除快,无副作用[61]。
在早期针对多肽受体的核素治疗中,90Y标记的奥曲肽类似物90Y-DOTATATE及90Y-DOTATOC因其高疗效在欧洲国家得到了广泛的应用。为增强治疗效果,第二代多肽受体核素治疗更加关注β放射核素,其中,177Lu标记的奥曲肽类似物,尤其是177Lu-DOTATATE在神经内分泌肿瘤的患者中具有高灵敏度及更长的无进展存活期[62]。Paganelli等2021年进行了177Lu-DOTA-奥曲肽Ⅱ期临床实验,观察治疗组的平均无进展存活期(PFS)和整体存活期(OS)。43例晚期胃肠道神经内分泌瘤,其中,男28例,女15例,平均追溯期为118个月,接受治疗剂量为18.5 GBq的患者的平均PFS为59.8个月,OS为71个月;接受治疗剂量为27.5 GBq的患者的平均PFS为59.8个月,OS为97.6个月;均未发现血液及肾毒性。临床2期研究显示,177Lu-DOTA-奥曲肽的多肽核素靶向治疗对晚期胃肠道神经内分泌瘤安全、有效。当然,肿瘤大小、肝损伤和年龄等因素是后续研究观察的重点[63]。2020年Zang等在晚期神经内分泌瘤的治疗当中将177Lu-DOTA-EB-TATE的剂量逐步提高至每个疗程3.97 GBq,患者未出现明显毒副作用。每个疗程所采用的用药剂量为1.89 GBq及3.97 GBq时,较1.17 GBq时更有效[64]。2020年Arveschoug等应用177Lu-DOTA-TOC治疗神经内分泌瘤的患者时,在发生外渗后,重新用药,在用药1 d后药物重新分布到肿瘤及血液中,患者未出现不适及副作用[65]。2020年Zahid等采用177Lu-DOTATATE治疗复发性脑膜瘤患者静脉注射给药4次,200 mCi/次,每次间隔8周,未发现副作用;肿瘤体积从第一次用药时的98.3 cm3缩小到第二次用药时的91.2 cm3,在第三次及第四次用药时肿瘤体积稳定在93.4 cm3[66]。2020年Parghane等用177Lu-DOTATATE对SSTR-转移性骨髓甲状腺癌患者进行治疗时,平均PFS为24个月,OS为26个月;降钙素倍增时间大于24 h能够提高PFS和OS[67]。
3.3 177Lu标记的其他分子
虽然,PSMA为靶点的放射性诊疗始于PSMA抗体,但目前在临床上受到关注的是小分子PSMA靶向配体,如,177Lu-PSMA-617在2015年被报道后迅速得到了临床应用[69]。近两年,177Lu标记小分子的177Lu-PSMA靶向配体药物的临床研究集中在治疗转移性去势抵抗性前列腺癌(mCRPC)。2020年Maffey-Steffen用68Ga/177Lu-PSMA-617作为诊疗配搭使用进行临床评价,纳入32例mCRPC患者,对接受177Lu-PSMA-617治疗的患者进行全身闪烁扫描,将全身剂量与68Ga-PSMA-617 PET/CT的结果进行比较,结果显示,177Lu-PSMA-617治疗的患者可用68Ga-PSMA-617进行治疗效果评价[70]。鉴于177Lu-PSMA治疗mCRPC患者的过程中,需要对前列腺特异性抗原(PSMA)进行监测以判断治疗效果。通常情况下该监测从治疗后的第12周开始,由于间隔时间较长,对治疗效果缺少早期评价。同时,采用小分子68Ga-PSMA-11作为PSMA探针时,由于PSMA-11的配位位置的原因,无法与其他三价态的镧系金属配位,因此177Lu无法实现单一核素的诊疗一体化[71-72]。2020年Gafita等将对前列腺特异性抗原(PSMA)进行监测的时间缩短至6周,临床上未发现与实际的治疗指标出现明显的偏差,有利于提供较早期的177Lu-PSMA治疗评价指标的同时,实现了利用177Lu同时完成对治疗和抗原检测[73]。α放射源(100 keV/μm)相对于β粒子(0.2 keV/μm)具有高线性能量转化,在临床放射诊疗中受到青睐[74]。相比于低线性能量转化,高线性能量转化产生的辐射会对DNA双链造成不可修复的破坏从而导致细胞的死亡[75]。考虑到30%的mCPRC患者对177Lu-PSMA-617无反应或产生抗性,因此,引进α核素增强治疗效果。如早在2016年Kratochwil等就采用了225Ac-PSMA-617治疗对177Lu-PSMA-617产生抗性的患者,展现了α核素的高效性[76]。但采用225Ac/177Lu-PSMA-617对mCRPC患者进行两种核素的联合治疗时,患者容易产生口腔干燥等副反应。Khreish等提出了联合用药的解决方案,即低剂量225Ac-PSMA-617联合177Lu-PSMA-617减轻了口腔干燥副作用的严重程度[77]。同时为减轻副反应,分批次用药的方案也在临床上进行了探索,2020年Zang等发现对mCPRC患者逐步提高177Lu-EB-PSMA剂量,剂量为2.12 GBq时安全有效的达成治疗目的,未发现肝肾毒性[78]。
2020年Yadav等通过直观类比标度(VAS)、镇痛标度(AS)和ECOG评分作为指标,评价177Lu-DOTA-Zol在缓解肿瘤骨转移引起的骨疼痛方面的临床应用,患者的疼痛明显缓解,无血液、肝肾毒性[79]。177Lu-DOTA-Zol可作为安全有效的肿瘤骨转移引起的骨疼痛的缓解剂。临床研究发现,不仅对乳腺癌引发的骨转移有效,对肺癌、前列腺癌引发的骨转移均有效。在DOTA-Zol的应用中,采用177Lu代替68Ga优点是血液清除快,但由于放射性快速的从血液中清除,并由肾脏排出,造成肾脏的放射性积累偏高[47]。以上研究的汇总列于表1。
4 小结
本文对近两年177Lu放射性治疗药物的临床前研究及临床应用进行了综述。近两年来,对177Lu放射性治疗药物的关注主要包括:
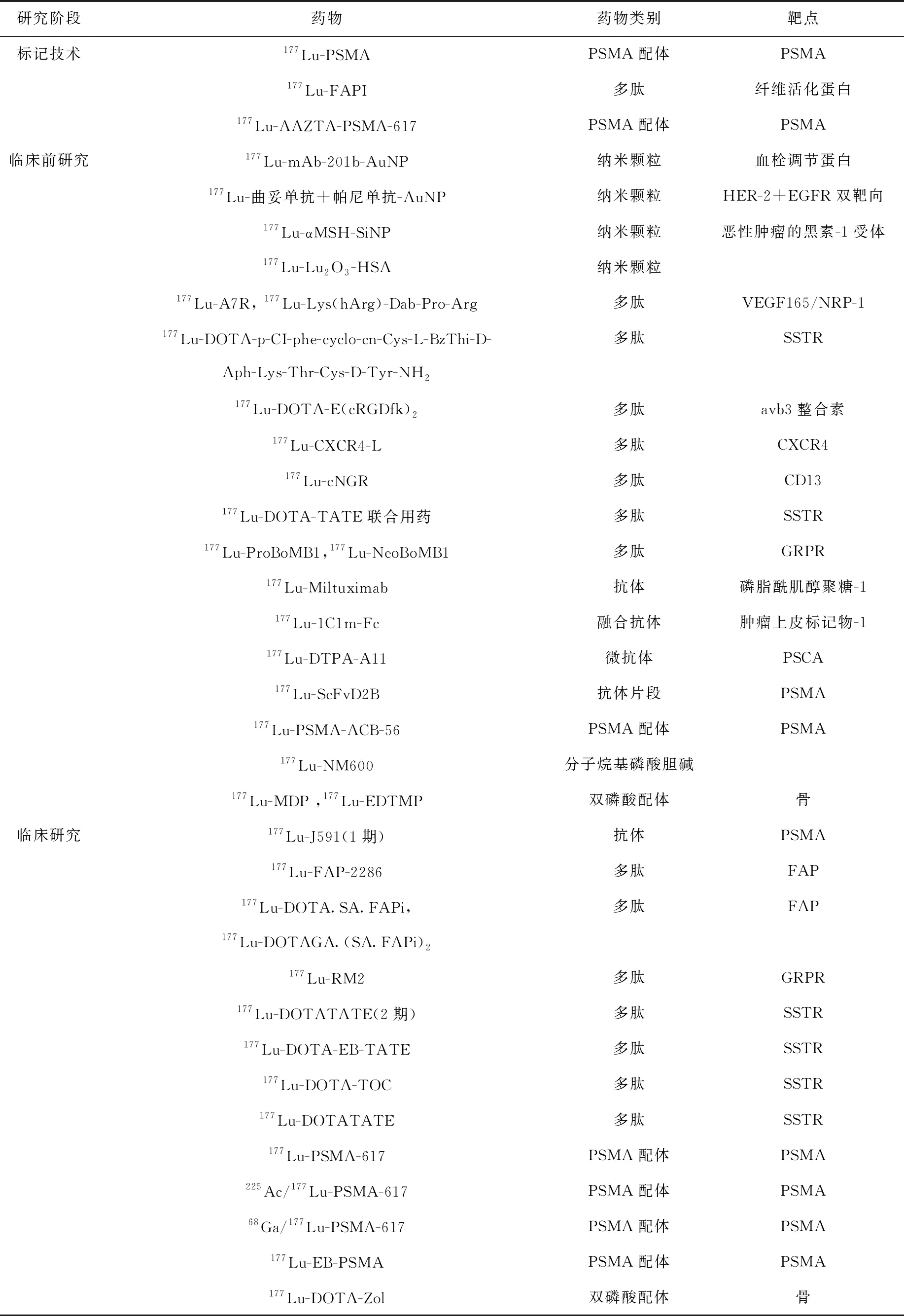
表1 2020—2021年177Lu放射性治疗药物汇总Table 1 Summary of 177Lu therapeutic radiopharmaceuticals in 2020 and 2021
1)177Lu药物药代动力学性能的改善,如采用抗体片段及微抗体取代抗体;2) 靶向性的提高,如双靶向177Lu放射性药物的设计;3) 标记多肽靶向治疗的多肽筛选,如受体激动剂与拮抗剂的对比,不同多肽结构的对比;4) 改进原有放射性药物的用药方式来增强疗效的临床探索及治疗效果评价的新方式方法,如联合用药的临床探索,患者剂量学评价以及诊疗结合判断治疗效果。
在177Lu放射性治疗药物领域,将涌现更多的研究成果,但找到生物性能更佳的多肽、单克隆抗体以及具有肿瘤靶向特性的新型小分子,将会在诸多研究成果中脱颖而出。
猜你喜欢
农村青少年科学探究(2022年4期)2022-07-29 14:40:50
保健医苑(2022年5期)2022-06-10 07:46:38
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
现代园艺(2017年13期)2018-01-19 02:28:09
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
环境与生活(2016年6期)2016-02-27 13:46:45
太空探索(2015年10期)2015-07-18 10:59:20
医学研究杂志(2015年7期)2015-06-22 11:01:01
药学与临床研究(2015年4期)2015-06-05 11:35:54
