
悬浮聚氯乙烯树脂微孔结构的调控
2022-06-21 01:50韩忠良汪海位马宝琪
中国氯碱 2022年5期
韩忠良,汪海位,马宝琪
(天伟化工有限公司,新疆 石河子832000)
悬浮法聚氯乙烯(PVC)树脂按照孔隙结构特点分为紧密型和疏松型, 疏松型树脂因其内部具备多孔结构,易于吸收各种加工助剂,便于在加工时被机械剪切力撕裂成小粒子从而加快塑化速度, 利于树脂在加工时快速完成塑化,并且达到一致的均匀性,这些特点使其在加工应用上的优势凸显, 普遍适用于各种加工形式的制品生产。 为此,PVC 树脂生产企业在生产助剂和工艺控制方面上下求索, 开发与应用新型助剂,全产业链工艺技术革新进步,保障了优质原料和辅料的提供, 为PVC 树脂微观品质调控、提升加工应用性能等创造了有利条件。悬浮法聚氯乙烯树脂内部丰富的微孔结构, 对其化学改性时的传质传热过程具有十分重要的意义。
悬浮聚合是PVC 树脂的四大聚合方式之一,是世界产能和产量最大的PVC 树脂生产工艺,悬浮疏松型PVC 树脂生产一般可通过分散剂、 搅拌强度、引发剂、 聚合温度、 转化率等工艺条件或配方的形式,对多孔结构进行控制。
1 分散剂调控
在悬浮PVC 宏观成粒过程中,搅拌和分散剂提供的剪切和保护作用, 使氯乙烯单体能够稳定悬浮在水相中。分散剂分子链上有亲水和亲油基团,常用的聚乙烯醇(PVA)类分散剂分子中既含有亲水性、高反应活性的羟基,又含有疏水的乙酰基团[1],随着醇解度的升高,羟基含量提高,亲水性增强,作用于水油两相界面,对胶体起到保护作用,减轻液滴间聚并形成的粘结颗粒。 许志东[2]等研究选用亲油性好的辅助分散剂,提高了树脂颗粒的疏松度和孔隙率。包永忠等[3]研究发现,随着PVA 醇解度增加,PVC 树脂的孔隙率降低,外形更加规整,表观密度增大;而复合使用时, 醇解度低的PVA 含量越高,PVC 树脂就越疏松,孔隙率越大。亲油性高的低醇解度聚乙烯醇,较多的乙酰基和较少的羟基嵌段分布于PVA 的分子链段上, 亲油性好的乙酰基能够迅速吸附在单体液滴表面甚至溶解于单体内部,羟基具有疏油性,当PVA 溶于单体液滴内部,其疏油的羟基能够继续在液滴内部形成阻隔, 在聚合物形成过程中减弱初级粒子之间的粘并,调节PVC 颗粒内部孔隙容积。
针对悬浮法PVC 树脂内部孔隙结构助剂调控,选取了两种50%左右醇解度但乙酰基分布有差异的PVA 作为辅助分散剂, 在相同条件下进行工业化对比试验,试验的树脂样品部分性能检测结果见表1。

表1 对比试验树脂性能检测
选用试验辅助分散剂PVA 生产平均聚合度800 的PVC 树脂, 树脂表观密度出现小幅下降,吸油率提升明显,同时“鱼眼”数下降,从结果分析,新型辅助分散剂的应用改变了PVC 的宏观和微观结构,使PVC 变得疏松多孔和易于塑化。 为比较PVC颗粒、内部亚颗粒和孔隙结构的变化,通过扫描电镜观察其外形结构和初级粒子聚集形态, 结果见图1和图2。 进行BET 比表面积测试,分析PVC 颗粒孔隙特征值,结果见表2。
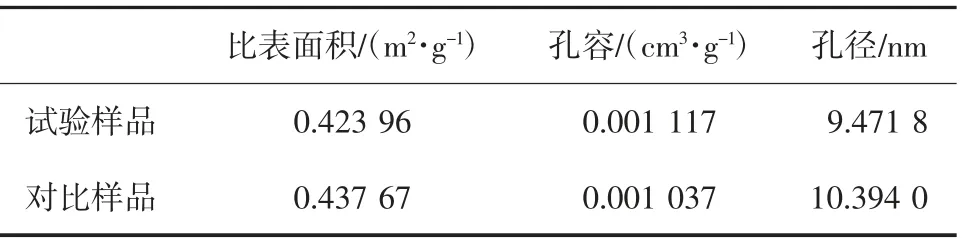
表2 不同辅助分散剂聚合产品BET比表面积特征值
对比两种PVC 颗粒的结构形态发现,图1 使用新型辅助分散剂的PVC 颗粒饱满, 表面结构规则、圆度较高,对表面裸露部分放大观察发现,初级粒子聚集程度高,大小较均匀。 图2 PVC 颗粒表面褶皱较多,且较多粘附的小粒子,表面裸露的初级粒子放大,初级粒子聚集度高,大小结构不均匀。 这可能是新型辅助分散剂的乙酰基和羟基结构分布特征的变化,减轻了内部不稳定的初级粒子凝聚粘并,提升了内部絮凝体强度, 在搅拌作用下, 破壳外溢几率下降,提高了颗粒内外平衡压力能力,降低了表面出现塌陷、褶皱结构。

图1 试验辅助分散剂生产PVC形态

图2 对比辅助分散剂生产PVC形态
两种样品BET 比表面积分析,试验样品的比表面积略小、平均孔径减小,孔容却出现上升,说明内部孔隙结构的丰度增大,均匀性更好,与增塑剂的吸收和电镜观察情况相吻合。
2 搅拌强度的调控
悬浮聚合需要搅拌提供动力, 形成流动场和剪切力, 将氯乙烯单体切割成小液滴, 搅拌强度对PVC 宏观成粒过程的影响已得到普遍认同,占晓强等[4]研究了搅拌转速对PVC 粒径的影响,认为在转化率10%以前改变转速对PVC 平均粒径影响较大,10%以后改变转速对平均粒径影响不大。 由此说明在低转化率阶段,PVC 颗粒结构尚未完全稳定,在搅拌剪切下形态可塑, 这一阶段颗粒内部氯乙烯富相和PVC 富相中均在发生聚合反应,随着转化率的升高,PVC 富相占主导时, 搅拌强度对宏观颗粒形态改变不大。 聚合初期搅拌强度对微观粒子的影响包括搅拌产生的离心力场、 自身所处环境重力场和浮力场等多重因素,使得PVC 微粒沉淀过程在三维立体空间完成,逐渐形成网络骨架结构,通过扫描电镜观察PVC 结构,PVC 颗粒及初级粒子形态结构见图3。
图3(a)中PVC 颗粒表面存在粘结的塌陷粒子,极可能为低转化阶段受搅拌作用, 搅拌剪切力突破分散剂的保护力, 溢出尚未成型的含聚合物和单体的固液混合物;图3(b)中裸露的初级粒子断面表现为不连续皮膜结构,从表面活性剂作用机理看,分散剂吸附不连续造成可能性不大, 可能是搅拌剪切将未定型态的PVC 颗粒撕裂形成断面,使初级粒子裸露;图3(c)中PVC 颗粒初级粒子断面周围能够清晰观察到初级粒子被皮膜覆盖的结构, 因而在悬浮聚合中调节宏观和微观颗粒形态, 分散剂与搅拌是协同作用的。

图3 PVC颗粒及初级粒子形态结构
3 引发剂的调控
PVC 不溶于VCM,PVC 链自由基增长到一定长度后,就有沉淀倾向[5],继续增长形成初级粒子核,链增长过程中液滴内部发生PVC 微纳粒子的沉淀、絮凝。 蔡启振等[6]研究发现,初级粒子长大和聚结达到一定程度形成网络结构,网络结构形成越早,强度越大,树脂颗粒的孔隙度也越大。初级粒子在转化率低时增长较快,随后平缓,高温下增长更快,加快早期的熔结,造成初级粒子增大、数量减少、孔隙减少、吸油率降低。高温提高了单个分子链增长的速率,引发剂分解的有效活性自由基形成多点引发, 短链PVC 长大形成的原始微粒不能单独成核,会再次絮凝形成初级粒子核。 原始微粒和初级粒子核等微观结构形成后,初级粒子核一经形成就开始成长,并吸附或捕捉来自单体相的自由基而增长、终止,聚合主要在PVC/VC 溶胀体中进行,不再形成新的初核。在一定聚合温度下,单个分子链增长的速率一定,表现的微观转化率可能几乎相同, 但引发剂分解成自由基浓度不同, 在单体中多点引发链增长反应形成宏观转化率是不同的, 可以认为在聚合初期尚未发生沉淀和絮凝前,每个点引发形成一条分子链,自由基的数量决定了初期形成短链数量, 数量越多形成的原始微粒越多, 再次絮凝形成的初级粒子核数量越多, 这样在后期初级粒子核形成的初级粒子数量更多,初级粒子均匀度更好,它们之间形成微孔结构越均匀和丰富。在聚合前期控制反应速率,避免过快形成致密的快速粒子,在加工时难以塑化成为“鱼眼”,甚至几乎无任何孔隙的玻璃珠。
引发剂的分解反应是一级吸热反应, 分解速率常数Kd与温度的关系符合Arrhenius 公式:
Kd=Ad×exp(-Ed/RT)
式中:Ad为频率因子;Ed为分解活化能;R 为通用气体常数;T 为绝对温度。
引发剂分解时间t 的浓度It与初始浓度I0之间关系为:
It/I0=exp(-Kd×t)
那么聚合反应时间t1到(t1+Δt)之间引发剂分解释放自由基浓度为2×(It1-It1+Δt)
按以上计算方法, 估算出38 ℃、57 ℃、65 ℃三种反应温度下, 所选配的复合引发剂体系分别为CNP+EHP、CNP+EHP、EHP+TMHP 体系, 聚合温度越高,在聚合初期引发剂分解释放的自由基量越小,聚合温度越低, 在聚合初期引发剂分解释放的自由基量越大。实际生产中,低温聚合所消耗的引发剂的物质的量要高于高温聚合,而低温聚合时得到PVC树脂吸油率和内部比表面积、孔容、孔径都普遍高于高温聚合的。 这样在聚合初期调控形成链引发剂自由基的数量可以达到调控微观成粒过程形成初级粒子数量的目标。
复配引发剂释放自由基,引发聚合反应,65 ℃反应聚合PVC 树脂内部结构见图4(a),57 ℃反应聚合PVC 见图4(b),38 ℃反应聚合PVC 见图4(c)。从图4 中可以清晰看出,图4(a)中裸露的初级粒子堆积紧密,初级粒子尺寸较大;图4(b)中裸露的初级粒子疏松,初级粒子尺寸大小不一,图4(c)中裸露的初级粒子密集、松散、尺寸小。 从微观成粒机理分析,反应初期,引发剂自由基活性中心引发聚合反应,小分子链长大、絮凝及形成初级粒子核,初级粒子核再长大沉淀, 开始反应时引发剂自由基数量决定了初核数量以及初级粒子数量,这可能是图4(c)中裸露的初级粒子细小、密集,图4(a)初级粒子少且尺寸大的原因。 如果引发剂活性自由基决定了初级粒子数量,那么温度则影响了初级粒子成长、沉淀速率,也可能影响到初级粒子聚集体的结构。

图4 不同量自由基引发的PVC树脂内部孔隙结构
4 聚合温度调控
聚合温度越高, 分子无规则运动越剧烈, 链引发、链增长速率越快,链转移终止的几率也增大,高温聚合得到的PVC 分子链较短。表面张力随温度的升高而降低,因此聚合温度越高,表面张力越小,单体越容易在水中分散和形成稳定的悬浮态。理论上,较小的表面张力更应得到疏松多孔的颗粒结构,不同聚合温度下生产的PVC 树脂吸油率和比表面积对比结果见图5。
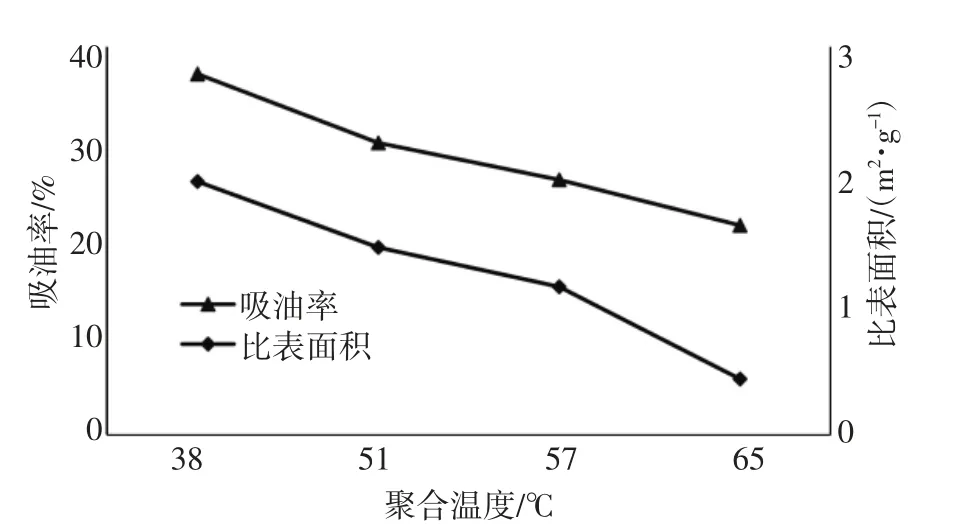
图5 不同聚合温度下PVC吸油率和比表面积
从图5 得出, 随着聚合温度的升高,PVC 的吸油率和比表面积值均出现下降, 聚合温度决定了引发剂的种类选择, 影响了引发剂分解活性自由基的效率,从而影响了聚合反应动力学速率快慢。 阚浩[7]等在研究氯乙烯本体预聚合动力学和成粒过程时发现, 在低转化率时, 搅拌对反应动力学几乎没有影响,提高聚合温度反应速率显著增大,且本体法与悬浮法动力学规律基本相似。 从38 ℃到65 ℃反应速率成倍增长,同时链转移、链终止速率也大大提高,因而得到短链PVC。然而在PVC 微观成粒的关键时期,温度影响了单个活性自由基引发的初核成长、絮凝和沉淀速度, 快速沉淀的初级粒子结构可能并不稳定,发生粘连概率增加,再加上成长速率大,初级粒子密度升高快,加剧了沉淀粘并结构的产生,进而影响初级粒子间空隙结构。反应热传导不及时,甚至发生早期熔结而形成粒子间的闭孔结构, 这一现象在图4(a)中能够得到印证。
5 转化率的调控
当初级粒子絮凝聚结形成三网结构后, 聚合反应主要集中在初级粒子的长大, 随着聚合反应的进行,初级粒子和其聚结体的直径和体积不断增大,颗粒内部的液相单体逐步参与反应变成固相, 由于PVC 不溶于VC 中, 而VC 却在PVC 中有相当的溶解量(30∶70),当转化率大于70%时,颗粒内部液相单体消失,大部分VC 单体溶胀在PVC 富相中并继续完成聚合。 梁斌等[8]在研究变温聚合工艺对PVC树脂性能的影响中, 采用后期升温反应, 得到PVC树脂表观密度上升,吸油率下降,认为是提高了单体转化率,填充了PVC 初级粒子间隙所致。 为考察转化率对PVC 内部孔隙率的影响,采用相同配方和相同聚合条件, 在试验聚合釜上进行不同转化率恒温聚合试验,结果见表3。

表3 不同转化率聚合PVC树脂特征
表3 反映出随着转化率的升高,PVC 的吸油率逐渐下降,表观密度逐渐升高,转化率83%以下时,难以塑化的“鱼眼”粒子数相差不大;达到90%时,“鱼眼”数显著升高。证明溶胀在PVC 内的氯乙烯继续完成聚合填充粒子间的间隙。 而随着氯乙烯富相的消失,反应速率越来越低,如继续延长反应时间获得较高的转化率,既不利于生产的经济运行,也不利于产品的质量控制。
6 结语
(1)选择合适的助分散剂,利用其特殊的官能团吸附在氯乙烯液滴上,保护单体相或聚合物,在聚合过程中能够提升PVC 树脂吸油率, 对调控PVC 孔容效果较明显;
(2)搅拌强度对PVC 宏观和微观调控主要集中于低转化率阶段,可与分散剂起到协同调控作用;
(3)引发剂加入种类和用量决定了聚合温度下分解活性基量的多寡, 大量活性自由基能够实现多点引发反应,总反应速率提升,温度则影响单个分子链反应速率的快慢, 实际生产中低温聚合反应消耗的引发剂有效自由基量高于高温聚合, 而低温聚合得到的PVC 树脂吸油率高,内部能见初级粒子粒径小、丰度高。
(4)转化率越高,PVC 树脂内部孔隙越小,控制合适的转化率既可以保障孔隙适中, 又能够获得较高的生产效率。
猜你喜欢
储能科学与技术(2022年2期)2022-02-19
磷肥与复肥(2021年9期)2021-11-09
气象与环境学报(2021年3期)2021-07-14
矿产勘查(2020年3期)2020-12-19
陕西科技大学学报(2020年3期)2020-06-15
三联生活周刊(2017年48期)2017-11-25
佛山陶瓷(2017年9期)2017-09-30
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
理科考试研究·高中(2014年8期)2014-10-17
