
气相色谱质谱法测定环境空气中苯并(a)芘的研究
2022-06-21 12:30郑贝贝吕玉新
河北环境工程学院学报 2022年3期
赵 超,郑贝贝,吕玉新
(1. 河南省万华环境检测有限公司,河南 新乡 453000;2. 河南省新乡生态环境监测中心,河南 新乡 453000)
多环芳烃(PAHs)是指分子中含有两个或两个以上并列苯环化学结构的一系列烃类化合物,是最早被人们认识的化学致癌物。 目前世界上的科学家已经检查出的500 多种主要致癌物中,有200 多种都是多环芳烃和多环芳烃的衍生物[1]。而苯并(a)芘就是多环芳烃中毒性最大的一种组分,是目前公认的三大一级致癌物(亚硝胺类、黄曲霉素、苯并(a)芘)之一,也是生活中常见的高活性间接致癌物和诱发基因突变的突变原。 经学者研究发现苯并(a)芘可诱发肺和结肠癌症[2-3]。当环境空气中的苯并( a) 芘浓度增加0.1 μg/100 m3时,肺癌的死亡率就会相应升高5%[4]。
1 空气中苯并(a)芘的来源及分析方法
环境空气中的苯并(a)芘来源广泛,主要来自于自然活动和人类活动。 大自然的自我活动如微生物的合成、自然火灾、活火山的喷发都会产生苯并(a)芘[5]。 人类的生产生活活动如工厂燃煤、生活燃煤、汽车尾气及食品烹饪制作[6]都会产生苯并(a)芘。 人类的活动是造成环境中苯并(a)芘污染的主要原因[7],特别是工业革命以后,人类对自然资源的消耗利用能力不断提高,生物化石能量燃料——煤和石油大量消耗,环境空气中苯并(a)芘的浓度不断升高。 环境空气中的污染物对人类健康的危害变得越来越不可忽视。 近年来,恶性肿瘤患者数量逐年呈上升趋势,据世界卫生组织估计,中国新增恶性肿瘤患者将占全世界新增患者的20%。
早期科学工作者们检测苯并(a)芘主要采用荧光分光光度法、紫外分光光度法,随着科学检测仪器的发展,气相色谱法、气相色谱质谱联用法、高效液相色谱法及液相色谱质谱联用法逐渐成为检测苯并(a)芘的主流方法[8]。 对环境空气中苯并(a)芘的检测,国内外普遍采用高效液相色谱法和气相色谱质谱法,通过采样器主动采样或用漏斗-瓶式收集器收集颗粒物沉降的采样方式采集空气中的苯并(a)芘,经萃取、浓缩、净化样品后,进入仪器分析。 还有学者通过空气中苯并(a)芘的逸度模型评估和预测环境空气中苯并(a)芘的浓度[20]。
本实验方法基于《环境空气和废气 气相和颗粒物中多环芳烃的测定 气相色谱-质谱法》(HJ 646—2013),通过采用手动进样脉冲不分流模式及增大进样体积以增大苯并(a)芘的进样量、质谱SIM 分析模式,大大降低了气相色谱质谱法测定苯并(a)芘的方法检出量和检出限。
2 材料与方法
2.1 仪器与试剂
气相色谱质谱仪,A91PLUS+AMD5(常州磐诺);色谱柱,AB-5ms(加拿大Abel Induseries);快速溶剂萃取仪,APLE-3500(北京吉天);固相萃取仪,SBEQ-CG1012(上海安谱);大气采样仪器,崂应2050 型智能空气综合采样器(青岛崂应);净化小柱,Florisil(1 g,6 mL)(上海安谱);氮吹仪,JHD-001(上海极恒)。 正己烷(HPLC 级)(上海安谱);丙酮(HPLC 级)(天津大茂);二氯甲烷(HPLC 级)(天津科密欧);无水硫酸钠(优级纯)(400 ℃烘干4 h);石英砂(20~100 目)(400 ℃烘干4 h);硅藻土(20~100 目)(400 ℃烘干4 h);玻璃漏斗;10 μL、25 μL、50 μL、100 μL、250 μL、500 μL 微量注射器;苯并(a)芘标准贮备液(上海安谱),浓度为2 000 mg/L;替代物标准贮备液(北京海岸鸿蒙):对三联苯D14,浓度为2 000 mg/L;内标贮备液(北京坛墨):苝D12,浓度为2 000 mg/L;超细玻璃纤维滤膜(400 ℃烘干4 h);玻璃棉。
2.2 仪器条件
色谱条件,进样口温度,280 ℃;进样方式,脉冲不分流进样,脉冲压力25 psi,脉冲时间1 min,在1 min 后分流,分流比60 ∶1;柱箱升温程序,80 ℃保持2 min,以20 ℃/min 速率升至180 ℃,保持5 min 后,再以10 ℃/min 速率升至320 ℃,保持5 min;柱内载气流量,恒定流量1.0 mL/min;进样量,2.0 μL。
质谱条件,离子源(EI 源);离子源温度280 ℃;传输线温度280 ℃;离子化能量70 eV;扫描模式-SIM 模式;溶剂延迟时间5 min。
2.3 仪器质谱性能检查
每次分析前,都应首先对质谱进行手动调谐,再将气相色谱质谱联用仪设定至分析条件,使其处于待机预运行状态,用微量注射器通过气相色谱进样口注入1.0 μL 十氟三苯基膦(DFTPP),运行设定的方法,得到其质谱图,其质量碎片的离子丰度应全部符合表1 中的要求。 否则有必要对气相色谱质谱仪进行维护:更换玻璃分流衬管、截掉5 cm 进样口端色谱柱以及清洗质谱的离子源。
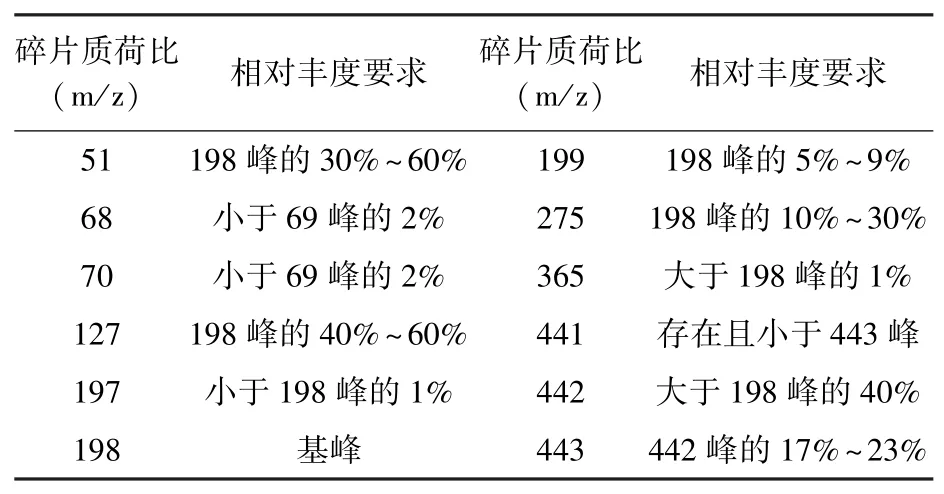
表1 DFTPP 调谐关键离子及离子丰度评价
3 实验过程
3.1 标准溶液的配置及曲线的绘制
用微量注射器分别移取苯并(a)芘标准贮备液和替代物贮备液500.0 μL 于10 mL 容量瓶中,用正己烷定容,混匀,配置的苯并(a)芘及替代物标准中间液各组分浓度均为100 μg/mL。 取上述标准中间溶液1.00 mL,用正己烷定容至10 mL 容量瓶中,混匀,各组分浓度为10 μg/mL。 移取内标贮备液200.0 μL 于10 mL 容量瓶中,用正己烷定容,配置浓度为40 μg/mL 的内标使用液。 按表2 配置0.025 μg/mL、0.050 μg/mL、0.100 μg/mL、0.200 μg/mL、0.500 μg/mL、1.000 μg/mL 系列标液于2 mL 棕色样品瓶中。

表2 标准溶液配置
按照仪器条件(2.2),从低浓度到高浓度依次用微量进样针进样分析,进样体积为2.0 μL。 以目标化合物浓度和内标化合物浓度比值为纵坐标;以目标化合物的定量离子响应值和内标化合物定量离子响应值的比值为横坐标,绘制校准曲线,如表3 所示。 图1 为标准色谱图。

表3 校准曲线
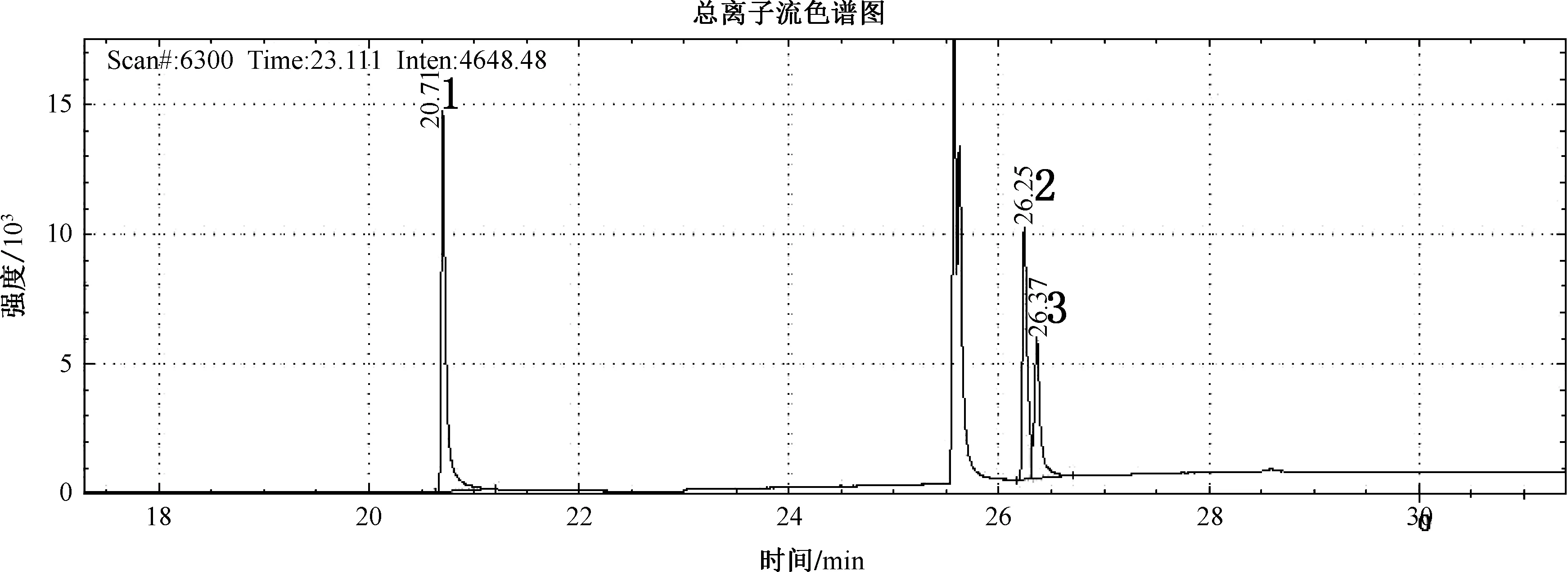
图1 苯并(a)芘标准样品的总离子流色谱图
3.2 样品的萃取
本实验用快速溶剂萃取仪提取样品中的苯并(a)芘。 使用的萃取溶剂为正己烷/丙酮(1 ∶1)溶液。 其他萃取条件如下:加热温度,100 ℃;萃取压力,10 MPa;加热时间,300 s;静态萃取时间,300 s;淋洗体积,60%;氮气吹扫时间,60 s;循环次数,2 次;清洗时间,30 s。
3.3 样品的浓缩
将提取液逐步转移至氮吹管中,干式氮吹仪的温度控制在35 ℃以下,调节氮吹流量使液面有轻微波动(避免形成气涡),浓缩时时常震荡氮吹管,并用正己烷少量多次冲洗氮吹过程中露出的管壁。 采样湿度较大的样品,在适当浓缩其提取液后会发现有水分存在。 在玻璃漏斗口径处填入玻璃棉,加入适量的无水硫酸钠,将浓缩后的样品倒入玻璃漏斗中,用15 mL 正己烷洗涤氮吹管后同样转入玻璃漏斗中,以除去样品中的水分。 若样品不需要净化,则将溶剂转化为正己烷后,浓缩样品至1 mL 以下,加入5.0 μL 内标使用液,定容至1.0 mL 后转移至棕色样品瓶,待分析。 样品需要净化时,将样品浓缩至2 mL 左右按3.4 净化。
3.4 样品的净化
将弗罗里硅土小柱(1 g,6 mL)固定于固相萃取仪,先后用5 mL 二氯甲烷溶液、10 mL 正己烷溶液活化小柱,淋洗速度控制在2 mL/min 左右。待小柱内充满正己烷后,关闭流速控制阀浸润小柱5 min,打开流速控制阀,弃去活化液。 在小柱筛板暴露于空气之前,关闭控制阀。 转移浓缩提取液至小柱内,用2 mL 正己烷,分两次清洗氮吹管后一并转入小柱内,打开流速控制阀同样使流速控制在2 mL/min,收集流出液。 在柱内溶液将要流尽、小柱筛板暴露于空气之前,关闭控制阀,加入2 mL 二氯甲烷/正己烷(1 ∶9)再次浸润5 min。 打开控制阀,用6 mL 二氯甲烷/正己烷(1 ∶9)洗脱小柱。 将净化后的样品液溶剂转换为正己烷后,浓缩至1 mL 以下,加入5.0 μL 内标使用液,定容至1.0 mL,转移至棕色样品瓶,待分析。
3.5 方法检出限
取7 个在马弗炉中400 ℃烘烤6 h 冷却后的超细玻璃纤维滤膜分别放入7 个萃取池中,将同样在马弗炉中400 ℃烘烤4 h 冷却后的石英砂和硅藻土混合装填于萃取池中,填装高度距离池口1 cm。 用微量注射器移取25 ng 苯并(a)芘分别加入萃取池中,按萃取方法(3.2)提取。 提取完成后,用氮吹仪小心浓缩至1 mL 以下。 分别加入5.0 μL内标使用液,定容至1.0 mL 后转移至样品瓶。 用气质联用仪按实验条件(2.2)逐个分析,其结果分别为0.031 μg/mL、0.030 μg/mL、0.033 μg/mL、0.029 μg/mL、0.034 μg/mL、0.035 μg/mL、0.035 μg/mL。
3.6 方法精密度和准确度
分别对空白加标量为0.050 μg、0.300 μg、0.800 μg 的样品进行6 次重复测定,计算其相对标准偏差,结果详见表4:
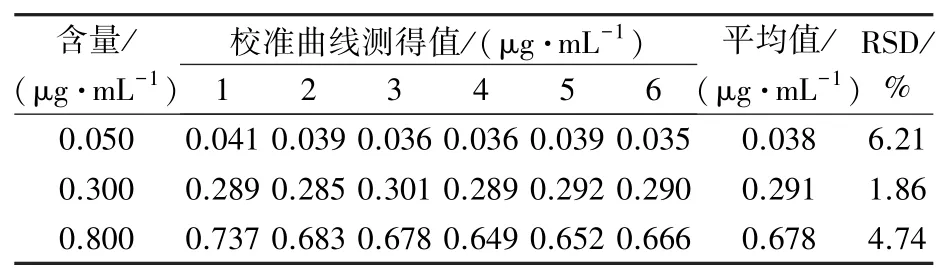
表4 方法精密度
在无苯并(a)芘污染的环境空气下,用崂应2050 型采样器以100 L/min 的速度采集60 min,分别采集12 个样品滤膜。 将12 个滤膜分为两组,分别对两组样品滤膜加标0.100 μg 和0.400 μg,经萃取、浓缩、净化、浓缩后加入5.0 μL 内标使用液,定容至1.0 mL 后上机分析。 计算其加标回收率,结果详见表5:

表5 方法准确度
3.7 实际样品的分析
组织采样人员对本地某防水材料公司厂界环境空气、某道路沥青生产单位的车间无组织空气、某炭素生产企业的设备旁无组织空气采样。 每个企业均采集3 个平行样品(100 L/min 采集60 min)。 两个样品作平行分析,第三个样品加标0.100 μg,计算RSD 及加标回收率,结果见表6:

表6 实际样品分析数据
4 方法与结果讨论
通过手动脉冲不分流进样和SIM 模式分析,改进了用气相色谱质谱联用仪检测环境空气中苯并(a)芘含量的方法,大幅度降低了苯并(a)芘的检出量。
4.1 样品预处理的优化
4.1.1 样品萃取溶剂的选择
我国《环境空气 苯并[a]芘的测定 高效液相色谱法》(HJ 956—2018)中采用的萃取溶剂为二氯甲烷。 从其加标回收结果可以看出,二氯甲烷对苯并(a)芘的提取效果很好。 但二氯甲烷对不锈钢材质的萃取池有一定的腐蚀性[9],且它还是2A 类致癌物,长期使用会升高脑部、肝脏和胆管等恶性肿瘤的患病风险[10]。 《环境空气和废气 气相和颗粒物中多环芳烃的测定 气相色谱-质谱法》(HJ 646—2013)用正己烷/乙醚(1 ∶1)作为提取液,但乙醚有较强的麻醉效能,不利于后续浓缩工作的开展。 通过实验发现单独使用正己烷时[11],多环芳烃的提取效率为70%~80%。 由于正己烷极性弱[12],单独使用正己烷萃取时的提取效率不够好。 综合诸多因素,本方法选择正己烷/丙酮(1 ∶1)作为提取剂。 丙酮极性较强,与弱极性正己烷搭配使用,可提高萃取效率。 且空气湿度较大时,采集的苯并(a)芘样品中可能存在水包油的情况,丙酮与水良好的互溶性可在提取时打破水包油,进一步提升萃取效率。
4.1.2 样品萃取方式的选择
大气中半挥发性有机物样品常用的提取方法有:索氏萃取、超声、超临界萃取、微波萃取、固相萃取和快速溶剂萃取[13]。 通过加标回收实验[14]对索氏提取、微波提取、超声波提取进行了比较,结果表明,索氏、微波、超声波的提取效率分别为97.2%,69.8%和92.2%。 将快速溶剂和超声波萃取两种处理方法进行了比较,结果显示两者萃取效率相近[15]。 不难看出索氏提取的效率最高,但其存在提取时间过长、有机溶剂用量大的缺点。超声波萃取所需的有机溶剂量同样大于快速溶剂萃取。 与索氏、超声、微波、超临界萃取方法相比,加速溶剂萃取有如下优点:有机溶剂用量少、萃取时间快、基体影响小、萃取效率高、安全性好[16-17]。另外快速溶剂萃取自动化程度高,不仅有利于减少提取过程中的偶然误差[18],而且无需实验人员长时间值守。 因此,本方法对样品的提取采用快速溶剂萃取法。
4.1.3 浓缩的优化
在浓缩时,要注意氮吹分压表的压力,压力太高、气流过大都会导致回收率降低。 丙酮和水可以互溶,所以提取的溶液直接用无水硫酸钠除水时,很难彻底除去水分。 而在适当浓缩后,低沸点的丙酮先挥发出去,此时除水会更彻底。 同时通过实验发现,除水时用15 mL 正己烷分两次淋洗加入样品后的无水硫酸钠,就可以保证有良好的回收率。 定容前要注意转换成与标液一样的溶剂,避免因为进样口的歧视效应造成分析误差。
4.2 萃取小柱的选择及净化的优化
实验选择用Florisil 柱净化提取液。 由于Florisil 硅羟基的作用力,极性弱的苯并(a)芘在柱上保留效果较弱,对极性的干扰物质保留能力强。不同于熊大伟等[19]的实验结果,本实验发现使用二氯甲烷/正己烷(1 ∶9)淋洗液8 mL,即能较好地回收全部苯并(a)芘,也不会因使用过多二氯甲烷而洗脱下部分极性物质。 净化过程中,注意保持2 mL/min 的洗脱速度,避免干扰物质尚未与填料相互作用或作用力小而被冲下来。 上样后浸润5 min 更有利于极性杂质与填料相互作用,从而避免干扰物质被洗脱下来。
4.3 进样方式的优化
分析实验中发现,在进同一个样品时手动进样比自动进样的响应值大。 脉冲不分流进样时,进样口的压力升至25 psi,在高压的作用下,更多的目标组分进入到色谱柱。 相对于普通不分流进样,脉冲进样时目标物组分特别是高沸点组分的响应值有大幅度的提高。 实验对比了进样量为1.0 μL、2.0 μL、3.0 μL 时的苯并(a)芘响应值,结果表明,进样2.0 μL 时,目标组分的信噪比最优。因此选择手动脉冲不分流进样,进样2.0 μL。
4.4 GCMS 分析的优点
GCMS 有双重定性的优点,可避免样品中杂峰的干扰。 且在预处理过程中添加替代物,通过分析样品中替代物的回收率可以检验该样品预处理的效果,是一种很好的质控手段。 样品的基体效应会干扰定量分析结果的准确性,在GCMS 分析方法中,内标校正可以消除部分基体效应,提高分析结果的准确性。 内标的添加还能在一定程度上消除因操作条件等的变化所引起的误差。 因此,用GCMS 法分析苯并(a)芘有着显著的优点。
4.5 影响测定结果的干扰因素
苯并(a)芘在环境中会发生氧化和光解[21],因此,样品和样品提取液要避免高温、阳光直射和接触氧化剂。 在GCMS 分析方法中,BbF 和BiF和苯并(a)芘有相同的碎片离子,可通过选择合适的色谱柱和优化柱箱升温程序,避免受其干扰。样品中苯并(a)芘含量相对较低,很容易受到采集运输、实验室环境、器皿和萃取溶剂的干扰。 用全程序空白和运输空白检查采样和运输过程中是否受到污染;通过实验室空白可以检查试验过程中是否受到污染;器皿使用后及时用清洗剂清洗,用甲醇润洗后放入烘箱烘干才可再次使用;购买的新批次的溶剂应做试剂验收,检查是否有本底值。 相较于普通不分流进样,脉冲不分流进样会缩短进样隔垫的寿命,可选择材质优良的隔垫且实验后及时检查以免其影响结果的准确性。
4.6 检出限的讨论
按公式计算7 次结果(3.5)的标准偏差为0.002 44。 按MDL=t(n-1,0.99)×S 计算方法检出限,当测定数为7 次时,其t(n-1,0.99)值为3.143。 计算可知当样品定容体积为1.0 mL 时,本实验方法的检出量为0.008 μg。 当采样体积为6 m3时,方法的检出限为1.4 ng/m3,检出下限为5.6 ng/m3。方法检出限与检出下限均低于大气污染物综合排放标准中苯并(a)芘无组织限值。 用本方法采集环境空气1 h,即可分析环境空气中苯并(a)芘的小时均值浓度。
《环境空气和废气 气相和颗粒物中多环芳烃的测定 气相色谱-质谱法》(HJ 646—2013)采用了SCAN 模式测定环境空气和废气中的苯并(a)芘,当样品定容至1.0 mL 时,苯并(a)芘的方法检出量为0.12 μg,远远高于本方法的0.008 μg。 标准中环境空气的推荐采样时间长达24 h,相较于本方法费时费力,不利于检测工作的快速开展。而当以100 L/min 的采样速度连续一小时采样6 m3时,其方法检出限为0.02 μg/m3,高于大气污染物综合排放标准中无组织空气中苯并(a)芘限值。
4.7 精密度和准确度
由表4、表5、表6 可知,本实验方法线性范围内的回收率为85.0%~116%,相对标准偏差为1.86%~10.9%。 表7 对比了本方法、HJ 646—2013 和HJ 956—2018 的精密度和准确度。 通过数据的比较可知,本实验方法的精密度和准确度均较好且稳定。

表7 精密和准确度的比较
5 结语
本方法使用了相对低毒性的正己烷/丙酮(1 ∶1)溶液作为提取液,避免大量使用对实验人员存在致癌和其他健康风险的有机溶剂。 通过技术手段改进了用气质联用仪检测环境空气中苯并(a)芘的方法,可以方便快捷地测定环境空气中苯并(a)芘的小时均值含量。 该方法满足大气污染物综合排放标准(GB 16297—1996)及环境空气质量标准(GB 3095—2012)的检测要求,且有较好的精密度和准确度。
猜你喜欢
少年文艺(2022年8期)2022-07-08
中国动物保健(2022年2期)2022-05-05
中国中医药信息杂志(2022年4期)2022-04-26
环境保护与循环经济(2021年7期)2021-11-02
环境保护与循环经济(2021年12期)2021-03-16
食品科学(2020年16期)2020-08-26
食品研究与开发(2020年9期)2020-05-08
汽车维护与修理(2020年17期)2020-03-10
中国经济周刊(2017年6期)2017-03-21
舰船科学技术(2016年1期)2016-02-27
