
Defect engineering for high-selection-performance of N2 activation over CeO2(111)surface
2022-06-20 06:22FngYunRishengSunLingFuGuozhengZho
Chinese Chemical Letters 2022年4期
Fng Yun,Risheng Sun,Ling Fu,*,Guozheng Zho,*
a Key Laboratory of Magnetic Molecules and Magnetic Information Materials Ministry of Education,The School of Chemistry and Material Science,Shanxi Normal University,Linfen 041004,China
b Institute of Environment and Energy Catalysis,Shaanxi Key Laboratory of Optoelectronic Functional Materials and Devices,School of Materials Science and Chemical Engineering,Xi’an Technological University,Xi’an 710021,China
ABSTRACT Nitrogen reduction reactions(NRR)under room conditions remain the challenge for N2 activation on metal-based catalysis materials.Herein,the M-doped CeO2(111)(M = Ca,Ti,V,Cr,Mn,Fe,Co,Ni,Cu and Zn)with oxygen vacancies,are systematically investigated by spin-polarized DFT+U calculations.We discuss briefly the situation of OVs on pure and reduced cerium,and we found that(1)doping TMs can promote the formation of oxygen defects,apart from Ti and V-dopant,(2)the O atoms are easier to escape connecting to M atoms than the ones of adjacent atoms connecting to the Ce(III),the value of OVs formation energies decrease as the TMs radius decrease.Also,our computational results show that Cr-doped,Mn-doped,Fe-doped,and Co-doped CeO2(111)adsorbs N2 strongly than the stoichiometric surface and other M-doped CeO2 surfaces with adsorption energies of-0.82,-1.02,-0.83 and-1.05 eV.Through COHP analysis,it is found that the predicted active sites have good catalytic performance.
Keywords:Nitrogen reduction CeO2(111)Defect Mechanism
As we all know,ceria(CeO2)is used in many applications,particularly as a catalyst in heterogeneous catalysis,photocatalysis,electro-catalysis[1,2].Generally,oxygen vacancies(OVs)play an important role in good catalytic performance,which comes from the interconversion between Cerium Ce(Ⅲ)and Ce(Ⅳ)oxidation states with the storage and release of 4f electronic orbits.It must also be mentioned that the formation of oxygen vacancies can change the distribution of the electrons on a cerium oxide surface,result in producing reactive and oxidative sites.Oxygen vacancies and Ce3+sites are the most active sites across the defective surfaces in the past studies[3–6].Esch and co-workers show that surface oxygen vacancy on CeO2is immobile at room temperature,but the linear cluster of these OVs,what is needed with the environment of higher temperature[7,8].And this makes the research on an activated mechanism of less oxygen vacancy more practical.
Apparently,there is no denying that increasing the number of the OVs is beneficial to enhance the nature of the catalysis,and there are many excellent reviews in the literature dealing with it.In experiments,Xie,Zhang and co-workers reported the exciting studies that CeO2nanorods with chromium(Cr),copper(Cu),and iron(Fe)doped can efficiently enhance the performance of electrocatalytic N2fixation to ammonia(NH3)because of increasing the OVs,in comparison to the pure CeO2nanorods[6,9–11].Also,Men and the laboratory team found that the synthesis of NiPt nanoparticles supported on CeO2nanospheres shows good catalytic performance for hydrogen generation from the alkaline solution of hydrazine at room temperature[12].In computational calculations,Luet al.also focus on CeO2(111)with doping manganese(Mn)and iron(Fe)for NO2adsorption and oxidation to NO3-by computational investigation.Also,some papers report that the interaction between CO molecules and samarium doped CeO2(111)surfaces promotes CO oxidation to CO2.Therefore,it indicates a good approach that doping the transition metals(TMs)to promote the formation of OVs.
It should be noted that the formation energy of oxygen vacancies in different layers and sites has a difference when CeO2dopes the TMs,and there is thereby an urgent need but it is still a significant value to discover and explain the contribution of the OVs with doped TMs to oxidation and reduction mechanism.To date,it is hard to summarize and understand regularly.In this work,our study demonstrates that(1)we built(3 × 3 × 1)supercell for studying the pattern about the formation of the OVs before and after doping the different TMs,with the analysis of the geometry,formation energy,and electronic structure,and investigated the relevant experimental evidence,what canbe prepared,to make sure that the research is meaningful[11,13–17].(2)Upon the ways of analysis with(1),we try to explain the regular formation to OVs with different TMs.(3)By analysis of the spin-charge density[18–21],we ensure the better layer doped site and the diffusion energy barrier according to the specific catalysis reaction to improve the efficient catalysts for application.
The main text of the article should appear here with headings as appropriate The calculations were performed at spin-polarized DFT+U approach using the Vienna ad initio simulation package(VASP)[22],which employs the generalized gradient approximation(GGA)with Perdew-Burke-Ernzerh of(PBE)functional.Also,we use DFT-D2 to describe the van der Waals bonds.For guaranteeing a good convergence of total energies,the plane-wave cut-off was 400 eV,the CeO2model uses a 20 ˚A vacuum layer to weaken the interlayer interaction,which uses the Brillouin zone sampled with 5 × 5 × 1 of k-points with allowing the convergence of total energy to set 0.01 eV,and the Part valence-electron configurations include Ce(5s,5p,6p,5d,4f),M(3d,4s)and O(2s,2p).Considering the 4f states of the reduced cerium atoms and 3d states of the third cycle transition group metals(Ca,Ti,V,Cr,Mn,Fe,Co,Ni,Cu and Zn)with the on-site Coulomb interaction,the value of the Hubbard U terms was used effectively[13,23–28],and the U value were determined by the report in other studies,which the value for Ce 4f was set to 5.0 eV,and we first predicted bulk lattice constant of the pure CeO2is 5.42 ˚A,which compared it and in agreement with the experimental value(5.41 ˚A)and the theoretical results(5.43 ˚A)[29–31].For the surface selection,we studied only the surface(111)among the low-index surfaces of CeO2(111),(110)and(100),which is the most stable[32,33].Considering the stability of the doped systems,the problem is investigated by calculating the defect formation energyEfdefined as[34,35]:
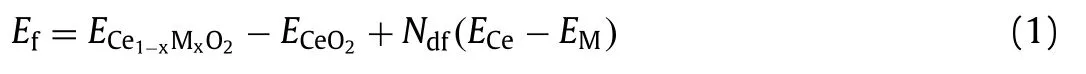
whereECe1-xMxO2is the total energy of the cerium with the dopant atoms,ECeO2is the total energy of the supercell-CeO2,Ndfis the number of dopant atoms,ECeandEMindicate the bulk energy per atom of Ce replaced and the dopant element M.With the instructions,a positive value is equal to the amount of energy to be doped,where the positive value indicates endothermic formation with larger the positive value,the more difficult it to form.
For investigating the formation energy of OVs on the CeO2(111)surface,the defect energyEvacis defined as following[36]:
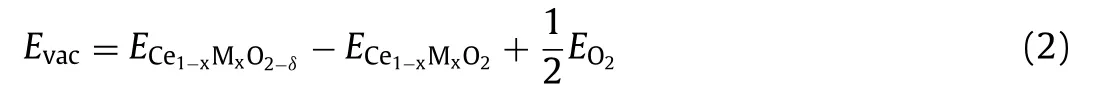
whereECe1-xMxO2-δ,EO2andECe1-xMxO2are the energy of the bulk in the presence of one oxygen vacancy,the energy of a gas-phase O2,and the bulk of surface Ce1-xMxO2,respectively.The definition shows that a positive value indicates endothermic formation and a negative value indicates exothermic formation.We use the same level to gain the formation energy of OVs in this work.
The adsorption energy(Eads)is defined as[37–39]:
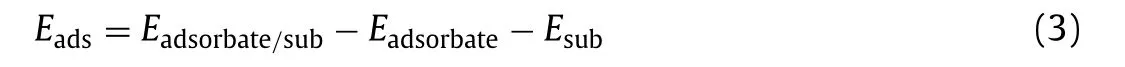
whereEadsorbate/sub,EadsorbateandEsubare the total energies of adsorbate-substrate,isolated adsorbate,and substrate system,respectively.And the negative value ofEadsindicates the better stable configuration and exothermic process.
The charge difference density(Δρ)plots were obtained from the difference between the charge density of the adsorbate surface system,the separated adsorbate and the bare surface:
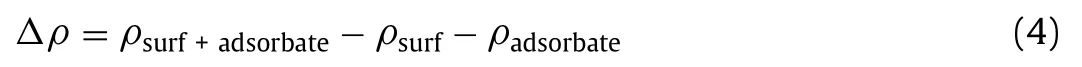
whereρsurf+adsorbate,ρsurf,ρadsorbateare the electron charge distributions of the M-doped CeO2surface with N2species,the isolated doped surface and the isolated adsorbed N2molecule,respectively.It is worth noting that the electron charge distributions of the isolated surface and isolated adsorbate origin from the optimized structures of the N2adsorbed systems,rather than the optimized free adsorbate and surface systems.
In the present study,the top and side views of the optimized CeO2were showed in Fig.1a.green,pink,red,brown,yellow atoms refer to surface oxygen,subsurface oxygen,the other oxygen sites,the doped site replaced Ce atom and the rest of the Ce sites,respectively.The circle place at O-top,Ce-top and Ce-O bridge sites indicates the location to possible adsorbed sites.It is worth noting that oxygen sites need to be studied all cited with the number 1-8.The DOS analysis(Fig.1b)shows that the orbital electronics of Ce(f)contributed between-1 eV and 0 eV,and Ce(d)orbital electronics contributed between-4 and-2 apparently.The bandgap energy value is 2.07 eV,which is well consistent with the value of 1.90 eV[31,40].Additionally,we calculated the formation of dopant energies of TMs,and theEfis 2.76 for Ca-doped system,corresponding to the calculated value of 2.20 eV by Jiaet al.[41],theEffor V-,Cr-and Mn-doped CeO2are 3.85,5.45 and 2.74 eV with theoretical research value of 4.46,5.31 and 2.36 eV by Wang[38].As suggested by Danny and co-workers,the valueEfof 13.52 and 11.47 eV for Cu and Zn doping,comparing with our calculated results of 8.16 and 12.52 eV[36,42].In the same ways,we calculated thatEffor Ti,Fe,Co and Ni doping is 0.47,9.65,11.09 and 5.80 eV,respectively.The results are basically same with the literatures.However,it is reasonable to get some deviation with different calculation methods(like different U values)and precision.
All defect models date of Ce–O and M–O bonds length and optimized stoichiometric CeO2surfaces were shown in Table S1(Supporting information).Herein,we present the partial and significant defect models of CrxCe1-xO2,CoxCe1-xO2,NixCe1-xO2and CuxCe1-xO2(Fig.S1 in Supporting information)for understanding the change of chemical bonding after the optimized calculation.The optimized pure CeO2(111)surface was also shown in Table S3(Supporting information),which has a length equal to 2.37 and 2.35 ˚A of the Ce–O bond,and the values were well consistent with the value of previous theoretical results[43–45].CeI and CeII are shown in Fig.S1.For the CrxCe1-xO2surface,there are short Cr-O bond distances of 2.19 ˚A(O5,O6 and O7)and long ones of 2.25 ˚A(O1,O2,and O3),and CeI-O4(CeⅡ-O8)of 2.32 ˚A.The optimized structure of Co-doped CeO2has shown the long Co-O2 bond distances of 3.45 ˚A and Co-O(O5 and O6)length of 2.81 ˚A.Also,the short Co–O(O1 and O3)bonds of 1.80 ˚A and Co-O7 bond of 1.91 ˚A,while CeI-O4 bond of 3.69 ˚A with CeII-O8 bond of 2.36 ˚A.From the NixCe1-xO2surface structure,we can see that the Ni atom has relaxed to the long Ni–O bond distances of 3.13(O1),3.24(O2),3.13(O3)and 2.05(O5 and O6)˚A,and one short Ni-O7 distance of 1.99 ˚A with CeI-O4 bond distance of 2.31 ˚A and CeII-O8 of 2.42 ˚A.
As shown in Fig.S2(Supporting information),it is worth mentioning the density of states to study the influence of doping TMs on the electronic structure of CeO2system.Figs.S2b and c show the states of LDOS and TDOS for defective cerium with OVS comparing to the pure CeO2,and no matter that the oxygen vacancy exists on the surface or sub,the models of CeO2-δpresent impurity states at the Fermi level from the pure cerium in spin up,which is the reason why the CeO2with OVs has an excellent performance in heterogeneous catalysis[46,47].According to the pure MxCe1-xO2(M = Ca-Zn)without OVs also have been observed in Figs.S2dm,we can see that the TDOS for Ca doped CeO2(Fig.S2d)reflects similar trend as one of the stoichiometric cerium without impurity state in the near of the Fermi level,which due to the proximity of atomic radii size between Ca(171 p.m.)and Ce(163 p.m.).The nature of TDOS of the Ti-,Cr-and Mn-doped CeO2represent the shifting with the impurity state at the plus value energy in spin up,above the Fermi energy pointed to the conduction band,which is shown in Figs.S2e,g and h.In addition,the DOS for Fe-,Co-,Ni-,Cu-and Zn-doped ceria shown in Figs.S2i–m reflect the unstable electronic properties due to energy states spin down near the Fermi level.In a word,it is not hard to see that the geometry and electronic structure of M-doped CeO2system have changed significantly,as the result of causing an electron-deficient region in cerium lattice,shifting the electrons from atoms neighboring sites to the less-electron-site and weakening the Ce-O or M-O bonds,by substituting Ce4+with TMs.Similar study has also been reported,recently[48–51].From the results,it is clear to know that atomic radii are an obvious effect on the structure of doped cerium,that larger or shorter than radii of Ce4+can usually make the larger geometric distortion and we conclude that doping way is good with potential for applicability(cheap on TMs of 4th-period elements)as a modality to create OVs.
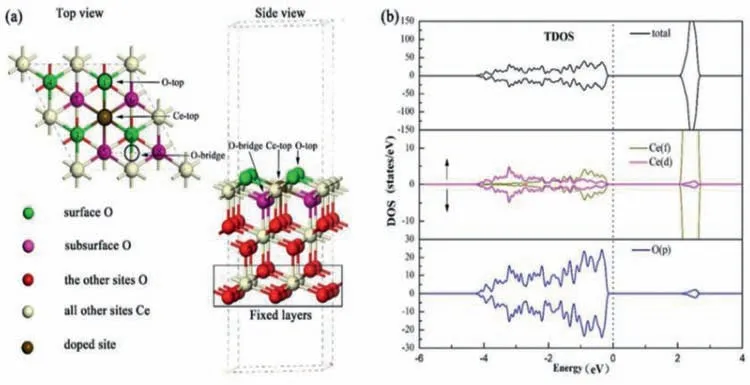
Fig.1.(a)Atomic structure of the CeO2(111)slab models from the top and side views.(b)The spin-polarized density of states(DOS)of the optimized stoichiometric ceria.The Fermi level is set at 0 eV.
To investigate the influence and law of dopants for oxygen vacancies,the situation of OVs on pure and reduced cerium should be discussed briefly.Table S2(Supporting information)shows the oxygen vacancies energies of MxCe1-xO2(M = Ca,Ti,V,Cr,Mn,Fe,Co,Ni,Cu and Zn)surfaces,which agree with the calculation by Michael Nolanet al.[41,51–54].Accordingly,the four types of oxygen vacancies(O1,O4,O5 and O8)around the doped positions,which are shown in Fig.1.The site 1(O1)and 3(O5)indicate the surface and subsurface OVs existing at the closest locations of M atoms,respectively.And the site 2(O4)and 4(O8)indicate the surface and subsurface OVs existing at next-neighbor sites,respectively.
As we already know,the formation energies for OVs(Evac)on the surface and subsurface of pure CeO2is 3.01 and 2.85 eV,and show the subsurface OVs are easier to generate[51,55–57].Importantly,the value ofEvacis getting lower close to zero,even negative once doping the TMs.However,theEvacof site 4(O8)on TixCe1-xO2-δsurface is 3.41 eV,which is larger than primitive CeO2without defects surface(Table S2).Based on the conclusions of geometric and electronic structure properties can remind us that the location of doped metal atoms with the oxygen atoms around the doped sites can leave and move away from the original positions,and therefore change the M-O bonds and adjacent Ce-O bonds.Along the y-axis and combined with the calculatedEvacin Fig.2,it should be noted that the value of oxygen vacancies formation energies of site 1 and site 5 are much lower compared with the OVs at site 4 and site 8 mostly.Specifically,for the Ca atom,the calculated oxygen vacancy formation energies are-0.78,-0.35,-0.55 and-0.75 eV of four types sites,respectively,and it was found that the radii of the Ca atom larger than Ce,which due to elongate the Ca-O bonds(Table S1),which Ca-O1 is 2.49 ˚A,Ce-O4 is 2.38 ˚A,Ca-O5 is 2.38 ˚A and Ce-O8 is 2.37 ˚A,conferring n-type of the dopants[58].For the Ti atom,theEvacof 1.77,2.71 and 3.41 eV correspond to the length with Ti-O of 1.91 ˚A(O1)and of for Ti-O.The Ni atom substitution on the cerium surface does facilitate the OVs formation of value-3.4,0.55,-3.19 and-2.92 eV whereas Ni atom is the most excellent one of all TMs this article studied to create OVs conferring p-type of the dopants.As well as the appeal TMs of Ca and Ni,the others also show the same nature,and the value of OVs formation energies decrease as the TMs radius decrease,which aimed at the same location for OVs sites to compare.
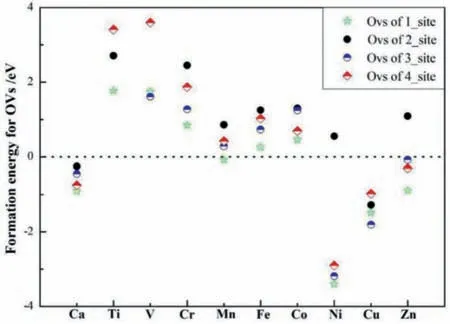
Fig.2.Comparison of calculated oxygen vacancy formation energies(Evac)for different sites for MxCe1-xO2(M = Ca,Ti,V,Cr,Mn,Fe,Co,Ni,Cu and Zn).Types of four OVs are defined in Table S2(Supporting information).
Generally speaking,by comparison of different OVs formation energies in Fig.2 and Table S3,we conclude that(1)doping TMs(Ca-Zn)way can promote the formation of oxygen defects,which would improve the significant redox performance,apart from Ti and V-dopant,which suppress the activation of Ce-O bongs nearby comparing with stoichiometric CeO2(111),(2)the O atoms connecting to M atoms are easier to escape than that of adjacent atoms connecting to the Ce,where TMs atoms insertion into the CeO2(111)surface,and(3)the value of OVs formation energies decrease as the TMs radius decrease,especially for the Ca,Ni,Cu and Zn-dopants,which can facilitate the oxygen vacancy formation spontaneously.
To contact with previous theoretical calculation[4,59–61],4f electronic orbits of Ce4+can capture two electrons to localize at reduced Ce3+cations sites,which occurred around the oxygen vacancy.In our studies,we performed specific electronic analyses to summarize and understand the influence of the TMs-doping in redox performance of CeO2(111)surface regularly,whose variable charge states of Ti,V,Cr,Mn,Fe,Co,Ni and Cu metals,and we found that there is no great charge accumulation on the surface of Ca-and Zn-doped models by comparison.
As shown in Fig.S3(Supporting information),the electronic structures of TixCe1-xO2-δwith one oxygen vacancy at 1,2 and 4 sites show different catalytic activity.Interestingly,the electrons not only localized at the Ce3+site but also M cations in Fig.S3c.Compare the three types of surfaces of electronic structures(Figs.S3a-c),it is easy to see that oxygen vacancy of site 4 at Ti-doped CeO2(111)subsurface is the most conducive to reduce the reaction,which exposing the most reduction sites.For TixCe1-xO2-δat site 4,the calculated Bader charge of Ti decreases to+2.16,with the formal charge of+3.Figs.S3d and e depict the LDOS results for TixCe1-xO2-δat site 4,wherein valence band is mainly contributed by Ce-f states in spin up with a minor contribution of O-2p states.While the conduction band is mainly a contribution of Ce-4f states,which far away from the Fermi level.The above results show that one of the 4f orbital electrons localizes at the Ce sites,and the other localize at the Ti cation site,which owns the reduction performance.Importantly,we find that the model of site 1 is the most stable by thermodynamic calculation and the diffusion energy is 1.64 eV from site 1 to 4.
Figs.S4a–c(Supporting information)reveal the electronic structures for V doped ceria of different sites 1,3 and 4,the electrons all localize at the Ce cation sites near the oxygen vacancy,and the sub-layer Ce cation is reduced shown in Fig.S4b.It is not hard to distinguish the best one of type site 4 by comparing the charge density and the location of the active sites.For VxCe1-xO2-δat site 4,the calculated Bader charge of V decreases to+2.23 with the formal charge of+3.In addition,VB is occupied by Ce-4f in spin up and O-2p,which far away of 0.97 and 2.01 eV from the Fermi level,respectively,and CB is occupied by Ce-4f states with minor O-2p and V-3d states,which closed to the Fermi level.The above results show that 4f orbital electrons localize at only Ce sites,which own the reduction performance.And the model of site 3 is the most stable by thermodynamic calculation and the diffusion energy is 1.85 eV from site 3 to 4.
For CrxCe1-xO2-δat different sites of 1,2,3 and 4,the electronic structures are depicted in Figs.S12a–d(Supporting information).The electrons all localize at the Ce cation and Cr cation sites near the oxygen vacancy,and Fig.S12c shows the sublayer Ce cation reduced.It is also easy to select the best one of type site 2 by comparing the number of active sites.With the analysis of the surface of Cr-doped CeO2(111)at site 2,the calculated Bader charge of Cr cation decreases to+1.92 with the formal charge of+2.Moreover,VB is occupied by Ce-4f states mainly in spin-up at the Fermi level,and the minor O-2p and Cr-3d states in spin-up which far away 0.13 eV from the Fermi level,and CB is occupied by Ce-f states closed to the Fermi level.The above results show that 4f orbital electrons localize at only Ce sites and Cr cation site with the reduction performance.And the model of site 2 is the most stable by thermodynamic calculation.
The reduced surfaces with different oxygen vacancies at sites1,2,3 and 4 of Mn-doped CeO2(111)are observed in Figs.S13a-d(Supporting information).The results show that none of the Ce atoms was reduced,which the electrons all localize at the Mn cation site with all sites.Fig.S5(Supporting information)indicates that the electrons occupying VB of site 4 have higher energy,which makes the reduced surface more active,as the Bader charge of this Mn cation was+1.52.Therefore,Figs.S13e and f depict the LDOS result of site 4 surfaces of Mn dopant with an oxygen vacancy,wherein,the valence band is mainly contributed by O-2p states with minor contributed by Mn-3d states with from the Fermi level at-0.67 eV,while major contribution in the conduction band of Ce-4f states far away from the Fermi level at 1.01 eV.The above cases show that one of the 4f orbital electrons localizes at the Ce sites,and the other localize at the Mn cation site owning the reduction performance.It is worthnoting that the model of site 1 is the most stable by thermodynamic calculation and the diffusion energy is 0.49 eV from site 1 to 4.
For FexCe1-xO2-δat different sites of 1,2,3 and 4,the electronic structures are depicted in Figs.S14a-d(Supporting information).The electrons all localize at the Fe cation of all sites(1,2,3 and 4),which exist in the site near the oxygen vacancy.Fig.S6(Supporting information)indicates that the electrons occupying VB of site 1 have higher energy across the Fermi level,which makes the reduced surface more active.It is also easy to select the best one of type site 1 by comparing the electronic structures.With the analysis of the surface of Fe-doped CeO2(111)at site 1,the calculated Bader charge of Fe cation decreases to+1.37.Moreover,VB is occupied by O-2p states mainly across the Fermi level,and CB is occupied by mainly O-2p states in spin up and minor contribution of Fe-3d states in spin-down across Fermi level.The above results show that 4f orbital electrons localize at only Fe cation sites with the redox performance as reporting that Fe cation site has excellent reductive nature of catalysis for ORR[62]by Zhonget al.,and showing the ability to fix nitrogen with doping Fe on MoS2/carbon cloth(CC)by Guoet al.[63–65].And it just so happens that the model of site 1 is the most stable by thermodynamic calculation.
Figs.S15a–d(Supporting information)reveal the electronic structures for Co-doped ceria of different site 1,2,3 and 4,the electrons all localize at the Co cation sites near the oxygen vacancy at 1,3 and 4,while the Ce and Co cation are reduced shown in Fig.S15b.It is not hard to distinguish the best one of type site 2 by comparing the charge density and the number of the active sites.For CoxCe1-xO2-δat site 2,the calculated Bader charge of Co decreases to+1.26.In addition,VB is occupied by dominating O-2p states and Co-3d states both in spin down,which far away of 0.13 from the Fermi level,and CB is occupied by Ce-4f states with minor O-2p states,which at the energy of+0.21 eV away from the Fermi level.The above results show that 4f orbital electrons localize at only Co sites,which own the reduction performance.And the model of site 2 is the most stable by thermodynamic calculation and the diffusion energy are 0.85 eV from site 1 to 2.
Also,Figs.S7a–d(Supporting information)have been observed in the electronic structures for Ni-doped ceria of different site 1,2,3 and 4,the electrons all localize at the Ni cation sites near the oxygen vacancy,while the Ni cation is reduced.And that the electrons occupying VB of site 4 have higher energy shown in Fig.S8.For NixCe1-xO2-δat site 4,the calculated Bader charge of Co decreases to+1.20.Furthermore,Figs.S7e and f(Supporting information)have shown the fascinating electronic structures as elaborated,wherein,mainly O-2p states and Ni-3d states in spin-up are found at Fermi level of–0.51 eV,while the conduction band is mainly contributed by Ce-4f states with minimal contribution of O-2p,Ce-5d and Ni-3d states at the deep CB,The above results show that 4f orbital electrons localize at only Ni sites,and the model of site 1 is the most stable by thermodynamic calculation and the diffusion energy is 0.47 eV from site 1 to 4.
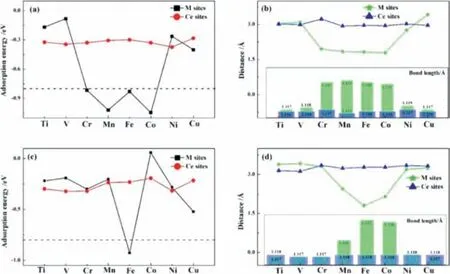
Fig.3.Calculated adsorption energies of N2 molecule of(a)N-N end-on and(c)N-N side-on on MxCe1-xO2-δ(M = Ti,V,Cr,Mn,Fe,Co,Ni and Cu)surfaces at different active sites.(b,d)show the distances from the surfaces of the adsorption sites and bond length for N2 molecular after the calculation is optimized.
Similar as the CrxCe1-xO2-δsurface,the oxygen vacancies at different sites of 1,2,3 and 4 demonstrate the electronic structures depicted in Figs.S9a–d(Supporting information),in which the electrons all localize at the Cu cation at the adjacency of oxygen vacancy.Comparing the TDOS of 4 types of Cu-doped CeO2(111)surfaces depicted in Fig.S10(Supporting information),indicates that the electrons occupying VB of site 3 have higher energy cross the Fermi level,which makes the reduced surface more active.So,to select the best one is type site 2 by comparing the electronic structures.With the analysis of CuxCe1-xO2-δthe surface at site 2,the Bader charge of Cu cation was calculated to be only+1.04 approximately with the formal charge of+2.Moreover,VB is occupied by Ce-4f states mainly in spin up at the Fermi level,and the minor O-2p and Ce-4f states cross the Fermi level,and CB is occupied by O-2p states and minimal Cu-3d states closed to the Fermi level of 0.21 eV.The above results show that 4f orbital electrons localize at only Cu sites with the reduction performance.And the model of site 3 is the most stable by thermodynamic calculation.
To verify the accuracy of charge analyses on MxCe1-xO2-δsurface and compare the active sits for reduction reaction with the predicted results.As shown in Fig.3,we built the models to carry out single-molecule adsorption simulations.Also,for Ti-,V-,Cr-,Mn-,Fe-,Co-,Ni-and Cu-doped CeO2with one oxygen vacancy,we built end and side-on ways to place N atoms at M cations and Ce3+sites for exploring the most stable adsorption structures in Figs.3a and c.Here,we test reduction performance by comparing the three parameters of adsorption energies,the distances between the N2molecule and active sites(M cations and Ce3+sites),and the bond length of the N2molecule.From the results in Figs.3a and c,we know theEadsof N2molecule are almost negative whether at M cations or Ce3+sites,and the negative value ofEadsindicates the better stable configuration and the exothermic process by the definition of adsorption energy.Specifically,by endon modes in Figs.3a and b,theEadsof N2molecule is-0.17,-0.09,-0.82,-1.02,-0.83,-1.05,-0.26 and-0.40 eV,respectively at M cations sites,and the adsorption energy are-0.32,-0.34,-0.33,-0.30,-0.29,-0.33,-0.37 and-0.28 eV,respectively at Ce3+sites.Interestingly,the N2adsorption is strong with the adsorption energy of-0.82,-1.02,-0.83 and-1.05 eV at M cations sites of Cr-,Mn-,Fe-and Co-doped models,which shows high activity for N2activation,corresponding to the bond length is 1.132,1.133,1.132 and 1.131 ˚A in Fig.S11(Supporting information).The bond length(1.122)is significantly longer than that of N2alone.For sideon modes in Fig.3c,theEadsof N2molecule is-0.21,-0.19,-0.29,-0.20,-0.93,0.06,-0.28 and-0.52 eV,respectively at M cations sites,and theEadsare-0.30,-0.32,-0.32,-0.23,-0.19,-0.21,-0.32 and-0.21 eV,respectively at Ce3+sites.Although most of the N2adsorption is weak,the Eadsis strong with the adsorption energy of-0.93 eV at Fe cations sites of the Fe-doped model,and it shows high activity for N2activation with the bond length is 1.137 ˚A in Fig.3d.
For understanding the activity of the M-doped CeO2-δ(M = Cr,Mn,Fe and Co)system,we introduced the concept of COHP.COHP(Crystal Orbital Hamiltonian Population)can be used to study the properties of local chemical bonds in periodic systems.The integral value ICOHP for the COHP below the Fermi level can be understood as the number of bonding electrons shared between two atoms.To some extent,it reflects the strength of the bond.Contributions to negative energy are bonding states,and contributions to positive energy are antibonding states.And we found the predicted active sites have good catalytic performance according to the COHP calculations(Fig.4)based on the previous studies that the excellent performance with NRR of TMs-doping-based catalysts can be considered to many occupied electrons of d-orbitals to release electrons to the antibonding orbitals of N-N triple bond[66–69].
From theab initioDFT calculations,we investigated the CeO2(111)with doping the different transition metals(Ca,Ti,V,Cr,Mn,Fe,Co,Ni,Cu and Zn)by studying geometric and electronic structure properties,and the influence of doping on the formation of four-types-OVs.Subsequently,we found that atomic radii and electronic structures with synergistic impact are two vital factors to modify and redecide structural states.We also take the specific mechanism for the change of Ce–O and M–O bonds after dopants to consideration,and we reported that the most active surfaces predicted are not necessarily the most stable at thermodynamics sometimes,which Ti-,V-,Mn-,Co-and Ni-doped CeO2-δwith relative active surfaces,the diffusion energy is needed of 1.64,1.85,0.49,0.85 and 0.47 eV,respectively.Importantly,the Cr-,Fe-and Cu-doped CeO2as well as the conditions of thermodynamics.
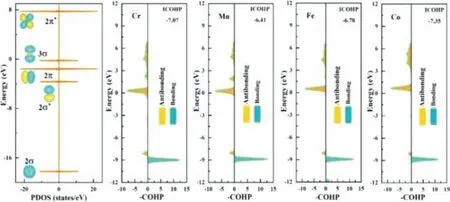
Fig.4.Calculated molecular orbitals the crystal orbital Hamilton population(COHP)of adsorbed N2 molecules on Cr-,Mn-,Fe-and Co-doped surfaces between N–N bonds via end-on configurations.
In summary,we reported two mechanisms of activation of MO and Ce-O bonds,which elongate the bonds with a larger size than Ce cation and less electron-transfer like Ca or shorten the bonds with smaller sizes and more electron-transfer like Ti-Zn.Surprisingly,when Ti-doped,the structure even suppresses the activation of Ce-O bongs nearby comparing with stoichiometric CeO2(111).In the end,the optimally reduced surfaces of doped TMs are TixCe1-xO2-δwith site 4,VxCe1-xO2-δwith site 4,CrxCe1-xO2-δwith site 2,MnxCe1-xO2-δwith site 4,FexCe1-xO2-δwith site 1,CoxCe1-xO2-δwith site 2,NixCe1-xO2-δwith site 4 and ZnxCe1-xO2-δwith 3.The key is that Cr-,Fe-and Cu-doped with one oxygen vacancy can obtain spontaneously the first rank of the surface,which is suitable for the reduction reaction.Finally,the results of adsorption energies and bond length with high activity for N2activation verify the predicted activesites for reducing power,which exhibits good performance for nitrogen reduction reactions(NRR)on the surface of Cr-,Mn-,Fe-and Co-doped cerium materials with oxygen vacancies.
Declaration of competing interest
The authors declare that they have no known competing financialinterestsor personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was funded by the Natural Science Foundation of China(Nos.21603109,21772152),the Henan Joint Fund of the National Natural Science Foundation of China(No.U1404216),the Scientific Research Program Funded by Shaanxi Provincial Education Department(No.20JK0676),the Transformation of Scientific and Technological Achievements Programs of Higher Education Institutions in Shanxi(No.2020CG032),the Cultivation Plan of Young Scientific Researchers in Higher Education Institutions of Shanxi Province,and the Fund for Shanxi ‘‘1331 Project”.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.09.003.
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis