
Meroterpenthiazole A,a unique meroterpenoid from the deep-sea-derived Penicillium allii-sativi,significantly inhibited retinoid X receptor(RXR)-α transcriptional effect
2022-06-20 06:21ChunLnXieDuoZhngKiQingGuoQingXingYnZhengBioZouZhiHuiHeZhenWuXioKunZhngHiFengChenXinWenYng
Chinese Chemical Letters 2022年4期
Chun-Ln Xie,Duo Zhng,Ki-Qing Guo,Qing-Xing Yn,Zheng-Bio Zou,Zhi-Hui He,Zhen Wu,Xio-Kun Zhng,Hi-Feng Chen,Xin-Wen Yng,*
a Key Laboratory of Marine Biogenetic Resources,Third Institute of Oceanography,Ministry of Natural Resources,Xiamen 361005,China
b School of Pharmaceutical Sciences,Xiamen University,Xiamen 361102,China
ABSTRACT A novel meroterpenoid,named meroterpenthiazole A(1),was isolated from the deep-sea-derived Penicillium allii-sativi.Its structure was established by extensive spectroscopic and computational methods.Meroterpenthiazole A bears a rare benzothiazole moiety in nature.Compound 1 significantly inhibited retinoid X receptor(RXR)-α transcriptional effect(KD = 12.3 μmol/L)through a novel binding mechanism.
Keywords:Deep-sea Fungus Penicillium allii-sativi Meroterpenoids Retinoid X receptor
Cyclohexane-linked drimane sesquiterpene are a class of meroterpenoid that combine a bicyclic drimane sesquiterpene unit with a cyclohexanone and cyclopentane moiety.Although the members of this molecules are relative rare in nature[1,2],the cyclohexane sesquiterpene have received significant interest from the synthetic community because of their attractive biological activities[3–6].Recently,the biosynthetic gene cluster of the epoxycyclohexenone macrophorins,MacJ,was identified[7].It was the first example of an integral membrane terpene cyclase that could cyclize terpenes through direct olefinic bond protonation.Interestingly,analysis of the secondary metabolite gene clusters of the deep-sea-derivedPenicillium allii-sativiMCCC 3A00580 revealed that a target gene clusterMerwas closely similar toMac(Fig.S1 in Supporting information).Accordingly,a systematic investigation was conducted on this fungus,which led to the isolation of a novel meroterpenoid,meroterpenthiazole A(1,Fig.1).We herein report the isolation,structure,biosynthetic pathway and bioactivity of this small molecule.
Meroterpenthiazole A(1)was obtained as a white powder.The molecular formula C26H34N2O4S was established based on its positive high resolution electrospray ionization mass spectroscopy(HRESIMS)atm/z471.2318[M+H]+(calcd.for C26H35N2O4S,471.2318),suggesting eleven degrees of unsaturation.The1H and13C nuclear magnetic resonance(NMR)spectra,with the aid of heteronuclear single quantum correlation(HSQC)spectrum,showed the presence of four methyls,one sp2and seven sp3methylenes,one sp2and two sp3methines,along with eleven nonprotonated carbons consisting of two carbonyls,seven vinylic and two aliphatic carbons.Moreover,two exchangeable protons were found.Since two carbonyls and nine olefinic carbons accounted for seven unsaturations,compound 1 was suggested to be a tetracyclic molecule.In the correlation spectroscopy(COSY)spectrum,three segments were deduced by correlations of H-25b to the 25-NH,H-2a to H-1a/H-3a,H-12aviaH-7b to H-6b and H-5,and H-12bviaH-9 to H2-11.In the heteronuclear multiple-bond correlation(HMBC),correlations originated from four methyls(H3-13/H3-14 to C-3/C-4/C-5,H3-15 to C-1/C-5/C-9/C-10 and H3-22 to C-18/C-19/C-20),three methylenes(H2-11 to C-16/C-17,H2-12 to C7/C-8/C-9 and H2-25 to C-26/C-24),one methine(H-21 to C-16/C-17/C-19/C-20),and the amino proton(25-NH to C-25/C-24)connected a benzene-linked drimane sesquiterpene and a glycine moiety(Fig.2).These two fragments,along with the remaining of one sp2non-protonated carbon(δC157.9 s)and two heteroatoms of N and S,were supposed to construct a unique benzothiazole meroterpene of 1.However,the limited HMBC correlations of 1 made it difficult to confirm its planar structure.Fortunately,the use of computer-assisted structure elucidation(CASE)programs can successfully determine structures for complex natural products,with the possible exception of proton-poor compounds[8–10].Accordingly,the 1D and 2D NMR as well as its HRESIMS spectroscopic data of 1 were analysed using the ACD/Structure Elucidator[11].As shown in Fig.2,the resulting highest-ranked structure(right)was identical to previously elucidated(left).Therefore,the planar structure of 1 was established as a novel meroterpenoid constructed by a sesquiterpene and benzothiazole.
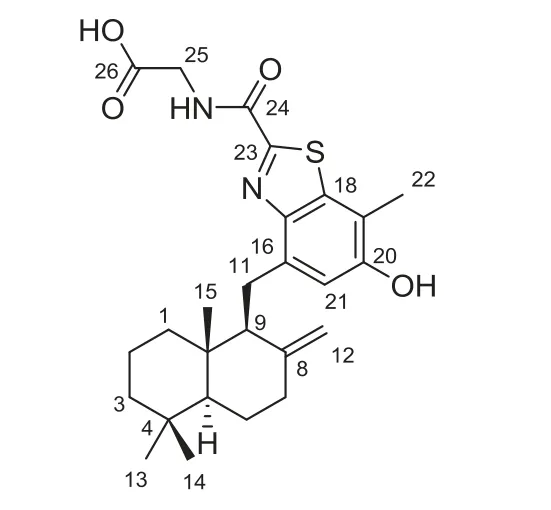
Fig.1.Chemical structure of meroterpenthiazole A(1).
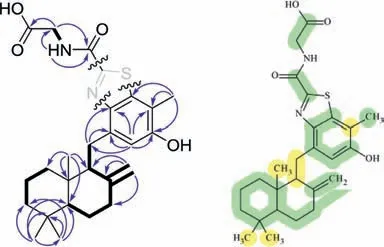
Fig.2.The key COSY(bold)and HMBC(arrow)correlations(left)and the analysis result of the suggested feasible structure(right)of 1 by ACD/Structure Elucidator programs(colored circles on the atoms display chemical shift differences:green denotes a difference of less than 3 ppm while yellow is between 3 and 15 ppm;deviations in neural network-based algorithm: dN(13C)= 2.322, dN(13C+1H)= 3.382).
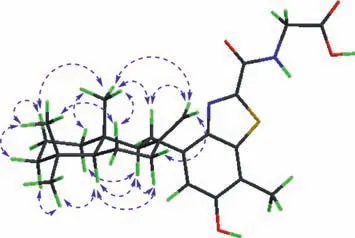
Fig.3.The key ROESY correlations of 1.
In the ROESY spectrum,correlations were found of Me-15 to H-1a/H-2a/H-6a/H-11a/H-21a,Me-14 to H-2a/H-3a/H-6a,H-21b to H-7a,and H-7b(δH1.94,td,J= 12.9,4.8 Hz)to H-6b/H-9(δH2.31 m)/H-5(δH1.23 m),suggesting Me-15 and H2-11 were on the same plane,which was opposite to that of H-5 and H-9(Fig.3).Therefore,the relative structure of 1 was determined as 5S*,9S*and 10S*,respectively.
For a verification,the quantum chemical calculation on the1H and13C NMR spectroscopic data was performed using the densityfunctional theory(DFT)method.Conformational analyses were performedviasystematic search algorithm at MMFF94 force field with RMSD threshold of 0.5 ˚A and energy window of 7 kcal/mol.Seventeen lowest energy conformers were obtained and were re-optimized at the B3LYP/6–31G*level in gas phase by the GAUSSIAN 09 program[8].The1H and13C NMR shielding constants of 1 were calculated with the GIAO method at MPW1PW91/6–311+G(2d,p)level in DMSO simulated by the IEFPCM model.The computational1H and13C NMR data(Table 1)were obtained by linear regression analysis[9],which agreed well with the experimental ones,with the correlation coefficients(R2)of 0.9930 and 0.9982,respectively(Fig.S4 in Supporting information).
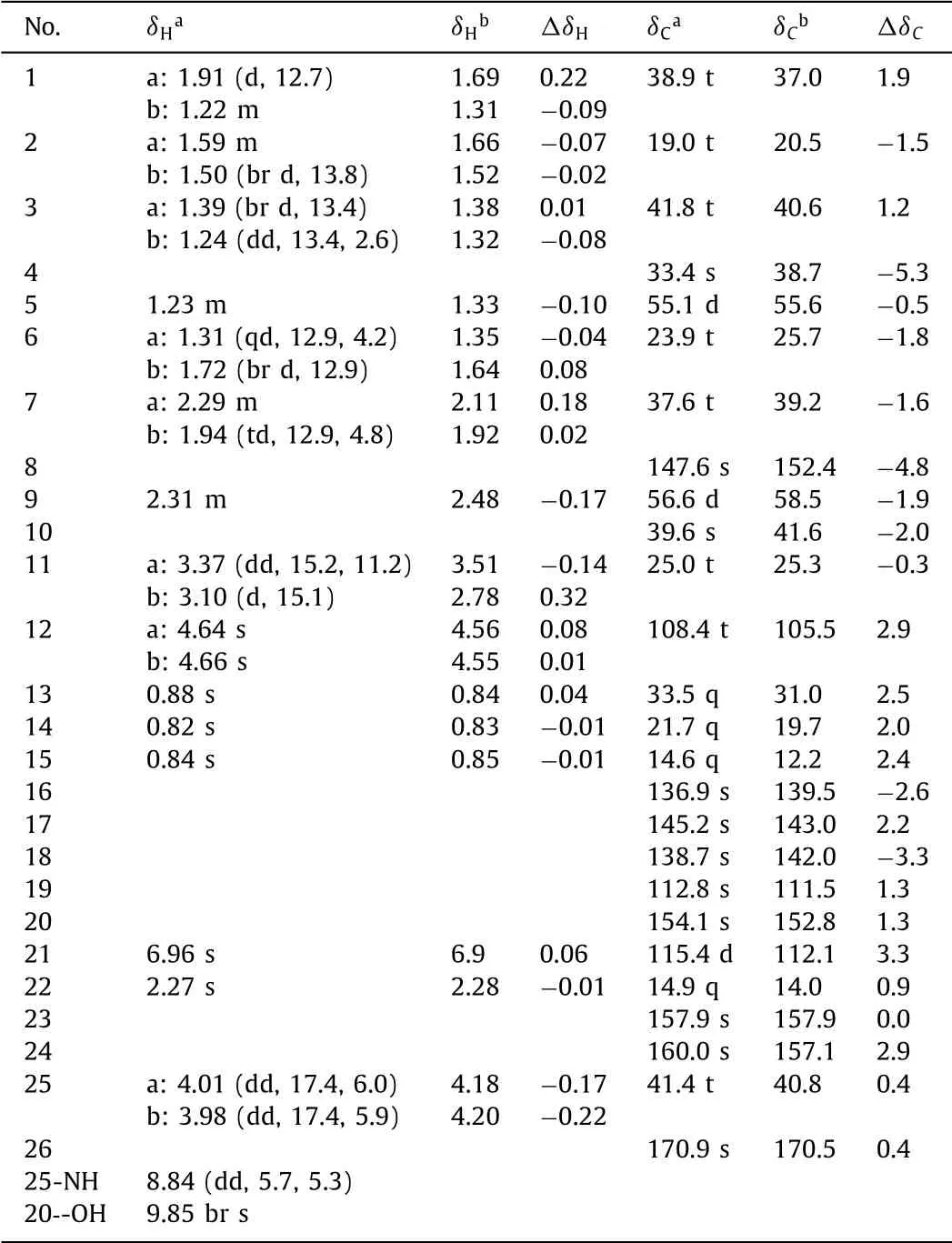
Table 1 The experimental 1H,13C and the calculated 13C NMR spectroscopic data for 1 in DMSO–d6(J in Hz).
To assign the absolute configuration of 1,a theoretical calculation of the electronic circular dichroism(ECD)was conducted using the GIAO method.Structures were obtained directly from previous optimizations.The calculations were executed in methanol with IEFPCM model using time-dependent density functional theory(TD-DFT).As shown in Fig.4,(5S,9S,10S)-1(1a)showed the same Cotton effects as the experimental one,while opposite to its enantiomer 1b.Accordingly,the absolute configuration of 1 was determined,and named meroterpenthiazole A.Noteworthily,the benzothiazole derivates are very rare in nature.Compound 1 is a unique feature among known meroterpenoids,and the enzymatic formation of this moiety is worthy to be studied.
As validated in the biosynthesis of yanuthone D,the key intermediate,5-farnesyltoluquinol,might be synthesized by the partially reducing polyketide synthase(PR-PKS)MerA,the decarboxylase MerB,the P450 MerC,and the prenyltransferase MerG[12].Then it was catalysed by the terpene cyclase similar toMacJ[7].The novel benzothiazine moiety should be originated from cysteine addition to benzoquinone derivate followed by ring contraction,as proposed in the biosynthetic pathway of the well-known firefly luciferin[13].Final step by a putative glycine transferase then constructed the whole structure of 1(Fig.S2 in Supporting information).
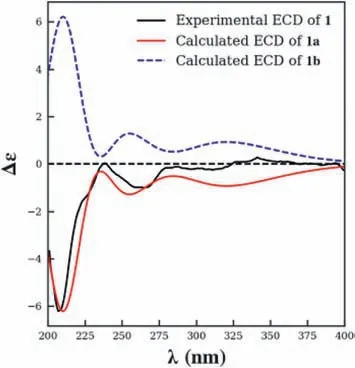
Fig.4.Calculated and experimental ECD spectra of 1 in MeOH.
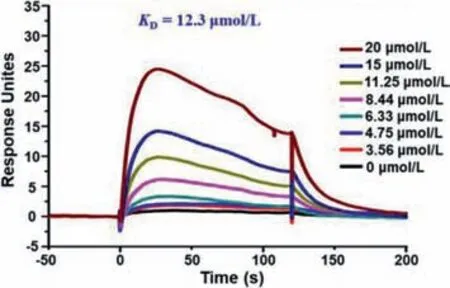
Fig.5.The SPR result of 1 binding to RXRα–LBD by Biacore T200.2.4.
Meroterpenthiazole A(1):white powder;MeOH);1H and13C NMR data,see Table 1;UV(MeOH)λmax(logε)205(3.77),205(3.16),205(3.48),205(2.84),205(3.43)nm;ECD(MeOH,1.0 mg/mL)Δε207(-1.77),238(+0.01),265(-0.29);HRESIMSm/z471.2318[M+H]+(calcd.for C26H35N2O4S,471.2318),493.2135[M+Na]+(calcd.for C26H34N2O4SNa,493.2137).
Retinoid X receptor(RXR)-αis an ideal target for antitumor drug design[14].Meroterpenthiazole A showed significant RXRαtranscriptional-inhibitory activities in a dose dependent manner(Fig.S6 in Supporting information).Using surface plasmon resonance(SPR)technology,meroterpenthiazole A was found to have direct interaction towards RXRα–ligand binding domain(LBD)withKDvalue of 12.3 μmol/L with the combination mode of fast association and fast dissociation process(Fig.5).
As reported,K-8008 could inhibit the TNFα-activated PI3K/AKT pathway to exert potent anticancer activity by mediating the interaction between a truncated form of RXRα(tRXRα)and the p85a regulatory subunit of PI3K[15,16].Since tetrazole group of K-8008 could act as biological electronic isostere of carboxyl and sesquiterpene grope of 1 shows the same hydrophobic effect to 4-isopropylbenzylidene in K-8008,they are structurally similar(Fig.S7 in Supporting information).Therefore,further investigations were conducted on the interaction mode with amino acid residues in RXRα–LBD by molecular docking in glide module.As shown in Fig.S8(Supporting information),meroterpenthiazole A could bind to the LBP of RXRα–LBD with the docking score of-5.752 kcal/mol,compared to-6.343 kcal/mol for K-8008.The terminal carboxyl formed H-bond lengths with Phe438,Phe439 of RXRα(2.5,1.9 ˚A),which were far shorter than that of K-8008(2.3,2.2 ˚A).While the drimane sesquiterpenes fragment can be bound to the hydrophobic pocket composed of Ile268,Ala271,Trp305,Phe438,Phe439 and Ile442 by hydrophobic force at 4.0 ˚A cutoff.Moreover,6-hydroxy in thiazole ring formed additional hydrogen bond(2.4 ˚A)with Cys269.The above results indicated that meroterpenthiazole A manifested a better binding conformation with RXRα–LBD.Therefore,same as K-8008,1 probably also binds to a tetrameric structure of the RXRα–LBD.
In conclusion,meroterpenthiazole A(1),an unprecedent benzothiazole meroterpenoid was discovered from the deep-sea-derivedPenicillium allii-sativiMCCC 3A00580.It could inhibit the RXRαtranscriptional activity by binding to RXRα–LBD,which may bring broad interests for chemical and biological synthetic chemistry.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors wish to thank Prof.Ying-Tong Di in the Kunming Institute of Botany for his constructive suggestions on the biosynthetic pathway of compound 1.The work was supported by the National Natural Science Foundation of China(No.22177143)and the COMRA program(No.DY135-B2–08).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.09.073.
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis