
Enantioselective total synthesis of(+)-vincamine
2022-06-20 06:21FanglinXueHengmaoLiuRuiWangDanZhangHaoSongXiaoYuLiuYongQin
Chinese Chemical Letters 2022年4期
Fanglin Xue,Hengmao Liu,Rui Wang,Dan Zhang,Hao Song,Xiao-Yu Liu,Yong Qin
Key Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry and Sichuan Province,Sichuan Engineering Laboratory for Plant-Sourced Drug and Sichuan Research Center for Drug Precision Industrial Technology,West China School of Pharmacy,Sichuan University,Chengdu 610041,China
ABSTRACT A catalytic asymmetric total synthesis of(+)-vincamine is presented.Key features of the synthesis include a Pd-catalyzed enantioselective decarboxylative allylation to form the C20 quaternary stereogenic center and a stereoselective iminium reduction to install the critical cis-C20/C21 relative stereochemisty.
Keywords:Indole alkaloid Decarboxylative allylation Stereoselective iminium reduction Natural product Total synthesis
The eburnamine-vincamine monoterpenoid indole alkaloids represent a large group of natural products found from the plants of genusHunteria,Vinca,andKopsia[1–8].(+)-Vincamine(1,Fig.1)is a unique member in this alkaloid subfamily that displays significant pharmacological activities and has been used as a peripheral vasodilator and nootropic agent in clinic[9–13].Additionally,other selected compounds belonging to this subfamily of alkaloids include(-)-eburnamonine(2),(-)-19-OH-eburnamonine(3),and unnatural(-)-20-epi-vincamine(4).Due to their significant pharmacological activities and limited natural abundance[14],the eburnamine-vincamine indole alkaloids became privileged synthetic targets for decades,which has resulted in a number of successful total syntheses[15–59].
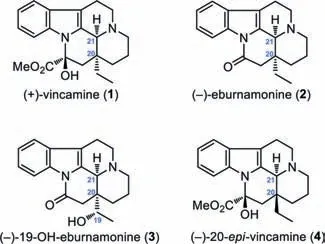
Fig.1.The structures of(+)-vincamine and related monoterpenoid indole alkaloids.
Despite the numerous efforts,two critical issues remain to be addressed toward an efficient enantioselective synthesis of the eburnamine-vincamine alkaloids.On one hand,catalytic asymmetric protocols for establishing the stereogenic chiral centers in these complex target molecules have been limited[36,42,46].The most known synthetic approaches to enantioenriched eburnaminevincamine alkaloids relied on resolution,chiral pool or chiral auxiliary methods[23–35,37-41,43–45,52,56,57,59].On the other hand,control of thecis-C20/C21 relative stereochemistry represents a key challenge in accessing the eburnamine-vincamine alkaloids[58].Recent endeavours in this research field have been particularly focused on solving the above-mentioned two issues.For example,Zhu and co-workers reported a conformation-directed cyclization process to selectively control thecis-C20/C21 stereochemistry in their total synthesis of(±)-eburnamonine[58].In 2019,the Trost group documented an enantioselective synthesis of C19-oxo eburnane alkaloids(e.g.,3)featuring a new Pd-catalyzed asymmetric allylation reaction[57].Recently,Chen,Tang,and colleagues developed an Ir-catalyzed asymmetric imine hydrogenation/lactamization cascade strategy to install thetrans-C20/C21 stereocenters(dr = 7.4:1)in their synthesis of(-)-4[59].During our preparation of this manuscript,Stoltzet al.published a catalytic asymmetric synthesis of(+)-eburnamonine((+)-2)with 3.4:1 dr in the formation of the C20/C21 relative stereochemistry[46].Our group previously described a photocatalytic radical cascade approach to(-)-vincamine((-)-1)[36].However,the key step(i.e.,6 to 7,Scheme 1)suffered from low diastereoselectivity(cis:trans= 3:2)of the C20/C21 relative stereochemistry.As part of our long-lasting interest in the total synthesis of complex alkaloid natural products[60–62],here we disclose our second-generation synthesis of(+)-vincamine(1)with excellent control of both the enantioselectivity and diastereoselectivity.
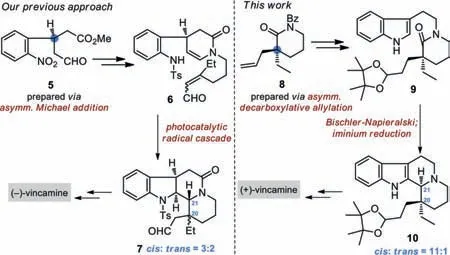
Scheme 1.Our two generations of asymmetric synthetic approaches to vincamine.
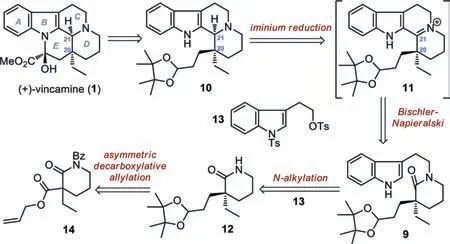
Scheme 2.Retrosynthetic analysis of(+)-vincamine(1).
Our retrosynthetic analysis of(+)-vincamine(1)is outlined in Scheme 2.According to known strategies,the E ring in 1 could be formed at the late stage of the synthesis through lactamization and subsequent rearrangement of 10.Preparation of the tetracyclic compound 10 could rely on a Bischler-Napieralski cyclization/iminium reduction sequence of amide 9,which would not only construct the C ring but also establish the configuration of the newly generated stereocenter at C21.We envisioned that introduction of a sterically hindered tetramethyl dioxolane group at the C20 side chain would greatly block the upper face of the iminium functionality in intermediate 11,thus securing a C20/C21cis-relationship after iminium reduction.In turn,amide 9 could be preparedvia N-alkylation of 12 using the indole fragment 13 as the electrophile.Finally,the quaternary stereocenter in 12 could be generated by an asymmetric decarboxylative allylation of 14 based on the Stoltz protocol[63].
The first challenge in the asymmetric synthesis of vincamine was to efficiently construct the all-carbon quaternary stereocenter at C20.Over the past decade,the Pd-catalyzed enantioselective allylic alkylation reaction has been a powerful tool for the construction of all-carbon quaternary stereocenters[64–67].Our total synthesis commenced with the Pd-catalyzed enantioselective decarboxylative allylation of racemic lactam 14(Scheme 3).Based on the slightly modified conditions(Pd2(pmdba)3(5 mol%),15(12.5 mol%),PhMe(0.1 mol/L),70 °C)of Stoltz’s report,we were able to obtain N-Bz piperidinone 8 with 93% yield and 98%eeon a gram scale[63].Ozonolysis of the terminal alkene in 8 in CH2Cl2at-78°C afforded aldehyde 16 with excellent efficiency(97% yield).Olefination of aldehyde 16 by Wittig reaction using methoxymethylene phosphonium chloride,followed by hydrolysis of the resulting methyl enol ether provided 17 in 87% yield over two steps.Subsequently,condensation of aldehyde 17 and pinacol in the presence ofp-toluenesulfonic acid delivered acetal 18(95% yield).Removal of theN-Bz group of 18 under basic conditions(LiOH·H2O,MeOH)gave the chiral lactam 12.
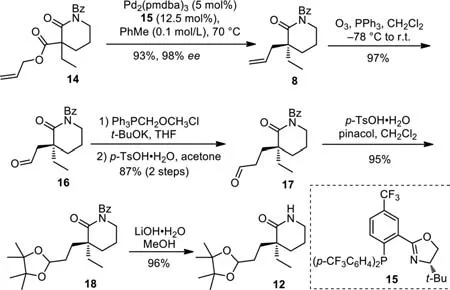
Scheme 3.Synthesis of the chiral building block 12.
Scheme 4.Total synthesis of(+)-vincamine(1).
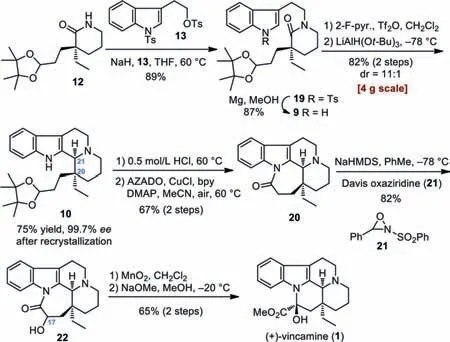
With the chiral building block 12 secured,we turned our attention to the linkage of 12 with an indole motif(Scheme 4).DirectN-alkylation of lactam 12 withβ-indolyl electrophiles proved challenging due to the instability of the latter under basic conditions.This problem was also encountered by Stoltzet al.in their recent synthesis of(+)-eburnamonine[46],who ultimately employed a stepwise approach to introduce the indole moiety.In our hand,after extensive experimentation,we were delighted to observe that tosylate 13 was a capable substrate to react with lactam 12 using NaH in THF at 60 °C to give the desired product 19 in 89% yield[68].Removal of the tosyl group at the indole Natom in 19 through treatment with Mg/MeOH yielded 9.At this stage,the key Bischler-Napieralski cyclization/iminium reduction sequence of amide 9 was explored.Subjecting 9 to 2-F-pyridine and Tf2O in CH2Cl2smoothly generated the iminium intermediate 11(Scheme 2)[69–72].After screening various reduction conditions(NaBHEt3,NaBH(OAc)3,NaBH3CN,DIBAL-H,L-selectride,LDBBA,H2/Pd-C,etc.),we found that the use of LiAlH(Ot-Bu)3at-78 °C furnished the tetracyclic product 10 with the optimal diastereoselectivity(dr = 11:1)in 82% yield over two steps.The above transformation(9 to 10)was easily conducted on a 4 g scale,which delivered the key intermediate 10 with requisitecis-C20/C21 stereochemistry in gram quantity with enhanced enantiomerical purity(99.7%ee)after recrystallization with acetone/water(4:1).Next,removal of the acetal group in 10 followed by oxidation of the resulting hemiaminal(structure not shown)according to Iwabuchi’s method[73]produced lactam 20,a known precursor to(+)-vincamine[32].Davis oxidation of 20 installed the C17-OH group and delivered hydroxyl lactam 22[59].Finally,MnO2-mediated oxidation of 22 and subsequent NaOMe-promoted rearrangement afforded(+)-vincamine(1)in 65% yield over two steps[26].
In conclusion,we have established a practical and concise approach to catalytic asymmetric total synthesis of the biologically important indole alkaloid(+)-vincamine in 14 steps and 16.2%overall yield.To a large extent,the current synthesis addressed the two major selectivity issues that have long puzzled the synthetic community by taking advantage of a Pd-catalyzed enantioselective decarboxylative allylation and a diastereoselective iminium reduction as key steps.Consequently,this synthetic strategy provides a general way to prepare other members and derivatives of the eburnamine-vincamine alkaloids possessingcis-C20/C21 stereocenters.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We gratefully acknowledge the National Natural Science Foundation of China(Nos.21991114,21921002 and 21732005)for financial support.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.09.032.
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis