
Is the metal involved or not?A computational study of Cu(I)-catalyzed[4+1]annulation of vinyl indole and carbene precursor
2022-06-20 06:21XioqinHeKngboZhongDnHengZhenZengHoNiRuopengBiYuLn
Chinese Chemical Letters 2022年4期
Xioqin He,Kngbo Zhong,Dn Heng,Zhen Zeng,Ho Ni,Ruopeng Bi,*,Yu Ln,b,*
a School of Chemistry and Chemical Engineering,Chongqing Key Laboratory of Theoretical and Computational Chemistry,Chongqing University,Chongqing 400030,China
b College of Chemistry and Institute of Green Catalysis,Zhengzhou University,Zhengzhou 450001,China
ABSTRACT The Cu(I)-catalyzed[4+1]annulation of vinyl indoles and a carbene precursor is a powerful method for constructing cyclopentaindole derivatives.Density functional theory(DFT)calculations were used to elucidate the mechanism and regioselectivity of this reaction.After Cu-assisted indole C3-alkylation,direct 1,5-annulation was favored over the Cu-assisted annulation pathway.Furthermore,the regioselectivity for 1,5-annulation was attributed to the generated five-membered-ring product being more stable than the three-membered-ring product from 1,3-annulation,which was the kinetically favored pathway.
Keywords:Carbene precursor Regioselectivity DFT calculations Cyclopentaindol C3-alkylation
Carbene[1–3]is a significant and valuable active intermediate in synthetic chemistry that can be considered a C1 source for the construction of new organic compounds[4–11].Carbene contains two unshared valence electrons and a neutral carbon atom,with a valence of two.Therefore,carbene can be considered an unsaturated carbon atom,with similar reactivity to other unsaturated molecules.In particular,carbene can be used as an unsaturated molecule to participate in cycloaddition reactions with other unsaturated molecules.A series of pioneering studies have been reported in this area[12–15].
Owing to its high reactivity,reactions using free carbene are usually difficult to control and obtain high selectivity from.Fortunately,introducing transition metals allows carbene reactivity to be regulated.The formation of metal-carbene complexes has led to extensive developments in carbene functionalization[16–23].Generally,Fischer-type metal-carbene complexes show electrophilicity because carbene donates one pair of electrons to the unoccupied orbital of late transition metals[24,25].Therefore,two possible reaction modes are proposed for Fischer-type metal-carbene complexes.In the mode A,the nucleophile attacks the carbon atom of the metal-carbene complex to afford a metal-carbon single bond,with electrophilic substitution(SE2)by another electrophile resulting in a bisfunctionalized carbene(Scheme 1,mode A)[26–30].Alternatively,we found that the nucleophilic addition intermediate can also dissociate from the metal species to afford a carbenion intermediate,with subsequent electrophilic substitution(SE1)occurring at theα-position of the enolate to give the product(Scheme 1,mode B)[31–32].
Recently,the Hu group reported a transition-metal-catalyzed functionalization of diazo compounds,in which Cu(CH3CN)4PF6was selected to catalyze the annulation of diazo compounds and vinyl indoles,affording cyclopentaindole derivatives P1(Scheme 2a)[33].We became interested in whether the annulation process was Cu-assisted and whether the vinyl indole could be considered as an unsaturated molecule or a nucleophile in this reaction.Therefore,the reaction mechanism and chemoselectivity were key problems in this reaction.In the present study,density functional theory(DFT)calculations were used to elucidate the mechanism and chemoselectivity of this reaction.
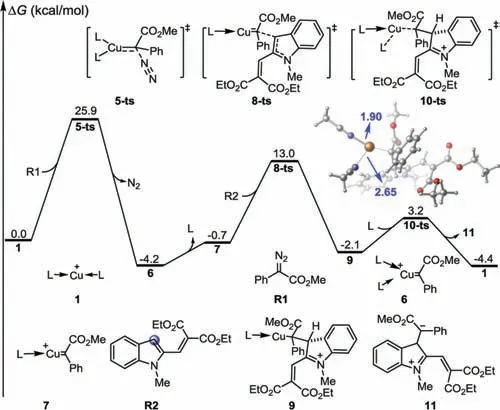
Fig.1.Gibbs energy profiles of the carbenation process.The values given by kcal/mol are the relative free energies calculated by M06-L/6–311+G(d,p)/SMD//B3-LYP/6–31G(d)(SDD for Cu)method in dichloromethane solvent.
All the density functional theory(DFT)calculations were carried out using the Gaussian 09 series of programs[34].Density functional B3-LYP[35]with a standard 6–31G(d)basis set(SDD[36]basis set for Cu)was used for geometry optimizations.Harmonic vibrational frequency calculations were performed for all stationary points to confirm them as local minima or transition structure and to derive the thermochemical corrections for the enthalpies and free energies.The M06-L[37,38]functional proposed by Truhlaret al.was used with a 6–311+G(d,p)basis set to calculate the single-point energies in the solvent,because it was envisaged that this strategy would provide greater accuracy with regard to the energetic information.The solvent effects were considered by single-point calculations in dichloromethane solvent base on the gas-phase stationary points with SMD solvation model[39–41].The energies presented in this paper are the M06-L calculated Gibbs free energies in dichloromethane solvent with B3-LYP calculated thermodynamic corrections.The noncovalent interactions(NCIs)[42–44]and electrostatic potentials(ESPs)were calculated at the B3-LYP/6–31G(d)(SDD for Cu)level.
As shown in Scheme 2b,possible pathways were accounted for during computational modeling of this reaction(Paths A and B).These pathways all started from cationic Cu(I)species I,which is carbenated by diazo compound R1,affording Fischer-type Cucarbene complex II.Intermolecular nucleophilic attack by the C3 position of vinyl indole R2 generates a zwitterionic intermediate III.In direct annulation pathway A,cleavage of the Cu-C bond release organic molecule IV with the regeneration of Cu(I)species I.Uncatalyzed 1,3-and 1,5-annulations of intermediate IV provide corresponding products VI and V.Alternatively,Cu-assisted annulation pathway B was also considered in our theoretical study.In this pathway,a 1,3-Cu shift generates Cu-enolate intermediate VII,which possesses a nucleophilic carbon for the subsequent 1,3-or 1,5-annulation.In this process,copper(I)is not eliminated until VI or V is formed as the final product.
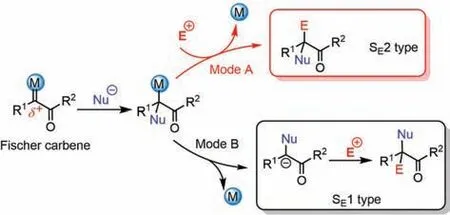
Scheme 1.Possible reaction modes for Fischer type metal-carbene complexes.
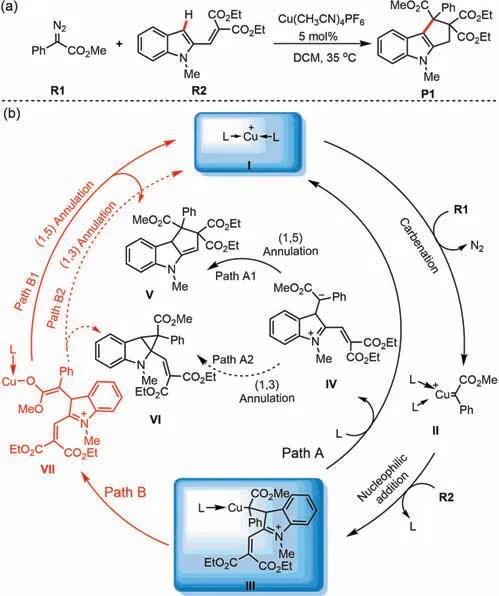
Scheme 2.(a)Cu(I)-catalyzed[4+1]annulation of vinyl indoles and diazo compounds and(b)its proposed catalytic cycle.
According to the proposed pathways,a detailed computational study was performed to investigate the mechanism of this Cu(I)-catalyzed annulation reaction.As shown in Fig.1,cationic copper(I)complex 1 was selected as the starting point for the free energy profiles,with two acetonitrile molecules selected as ligands(we consider the four coordination patterns for copper,see Fig.S3 in Supporting information for details).Cu-assisted aryldiazoacetate R1 denitrogenationviatransition state 5-ts affords Cu(I)-carbene complex 6,with a free energy decrease of 4.2 kcal/mol.The overall activation free energy for the carbenation process was 25.9 kcal/mol,which was the highest free energy barrier in the entire pathway.Therefore,carbenation was considered to be the rate-determining step of the catalytic cycle[45,46].Subsequent dissociation of one acetonitrile molecule afforded complex 7 with an increase in free energy of 3.5 kcal/mol.In the presence of dimethoxycarbonyl vinyl indole R2,C3 nucleophilic attack could occurviatransition state 8-ts to form zwitterionic intermediate 9.The chemoselectivity of nucleophilic addition was also investigated(see Fig.S4 in Supporting information for details).Ligand exchange with acetonitrileviatransition state 10-ts releases organic species 11 with the regeneration of active species 1(see Fig.S5 in Supporting information for details).
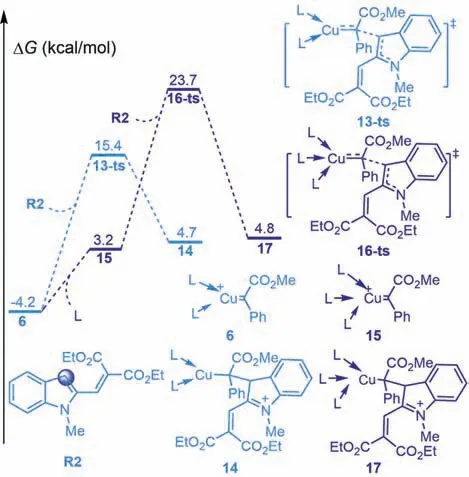
Fig.2.Gibbs energy profiles of the Cu(I)-carbene complex 6 and 15 to nucleophilic addition process.The values given by kcal/mol are the relative free energies calculated by M06-L/6–311+G(d,p)/SMD//B3-LYP/6–31G(d)(SDD for Cu)method in dichloromethane solvent.
In addition,we have considered the different coordination patterns for Cu(I)-carbene complex.As shown in Fig.2,C3 nucleophilic attack of metal-carbene 6 to dimethoxycarbonyl vinyl indole R2viatransition state 13-ts with an energy barrier of 19.6 kcal/mol to gives zwitterionic intermediate 14.This process is endergonic by 19.6 kcal/mol,which can be attributed to entropy loss.The relative free energies of transition state 13-ts were 2.4 kcal/mol higher than that of transition state 8-ts(Fig.1).In the other case,the coordination of two acetonitrile to the copper center results in formation of unstable intermediate 15 in an endergonic process with 7.4 kcal/mol free energy.Then C3 nucleophilic attackviatransition state 16-ts to gives zwitterionic intermediate 17 with an energy barrier of 20.5 kcal/mol.The relative free energies of transition state 16-ts were 10.7 kcal/mol higher than that of transition state 8-ts(Fig.1).Therefore,the paths involving the nucleophilic addition of intermediates 6 and 15 are unfavorable.
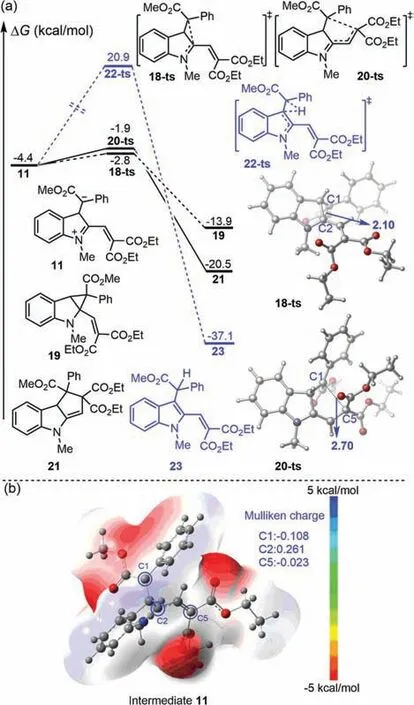
Fig.3.(a)Gibbs energy profiles of direct annulation.The values given by kcal/mol are the relative free energies calculated by M06-L/6–311+G(d,p)/SMD//B3-LYP/6–31G(d)(SDD for Cu)method in dichloromethane solvent.Optimized geometries of transition states 18-ts and 20-ts.Bond lengths are shown in Angstroms(˚A).(b)Electrostatic potential map and Mulliken charge for intermediate 11.
Having obtained key intermediate 11,direct annulation of this intermediate was considered theoretically(Fig.3a).DFT calculations found that the free energy barrier of the 1,3-annulation processviatransition state 18-ts was only 1.6 kcal/mol.However,generated cyclopropane-type product 19 was only 9.5 kcal/mol more stable than intermediate 11.This clearly showed that compound 19 was the kinetic product,which can be obtained reversibly from 11.Alternatively,the corresponding 1,5-annulationviatransition state 20-ts gave an energy barrier of 2.5 kcal/mol,but as generated cyclopentene product 21 was 6.6 kcal/mol more stable than 1,3-annulation product 19.Therefore,compound 21 was the thermodynamic product.The entire annulation process can be descripted as a reversible 1,3-annulation of intermediate 11 to provide intermediate 19,which undergoes the reverse process to regenerate intermediate 11.Subsequently,1,5-annulation finally yields thermodynamic product 21.In this case,the overall activation free energy for the generation of 21 from 19 was only 12.0 kcal/mol.Therefore,compound 21 was observed as the major product experimentally.The rearomatization of intermediate 19 was also considered theoretically(see Fig.S6 in Supporting information for details).Although the free energy barrier of an intramolecular 1,2-hydrogen shift from intermediate 19 was only 25.3 kcal/mol,considering the generation of 21,the overall activation free energy for the formation of alkylated vinyl indole product 23 was up to 41.4 kcal/mol.The electrostatic potential(ESP)of complex 19 is shown in Fig.3b,clearly elucidating its character.In this structure,negative charge was mostly located at the C1 position,which could act as a nucleophile,while positive charge was mostly located at C2,which exhibited electrophilicity.Therefore,the formation of a C1-C2 bondvia1,3-annulation is kinetically favorable,but the generated three-membered ring is thermodynamically unfavorable.Interestingly,C5 also exhibits weak electrophilicity,and can bond with C1viathe corresponding 1,5-annulation.Generation of the five-membered ring is thermodynamically favorable.
The Cu-assisted annulation was also considered theoretically.As shown in Fig.4,when intermediate 9 is generated by nucleophilic addition,a 1,3-Cu shift can occurviatransition state 24-ts with a barrier of 9.1 kcal/mol.Generated intermediate 25 possesses a nucleophilic enolate carbon,which can undergo corresponding annulation.Similar to direct annulation(Fig.3a),the free energy barrier of the 1,3-annulation processviatransition state 26-ts was only 2.5 kcal/mol,leading to the formation of kinetic product 19.The corresponding 1,5-annulationviatransition state 27-ts had an energy barrier of 7.5 kcal/mol,affording thermodynamic product 21.The electrostatic potential(ESP)map of complex 25 clearly showed that the presence of Cu decreased the nucleophilicity of the enolate moiety(Fig.4).Therefore,a higher energy barrier was observed in the 1,5-annulation process.
Interestingly,when asymmetric 1,3-dicarlbonyl compounds was used as substrate,the 1,5-annulation exhibited diastereoselectivity.For example,when R3 was used as the 1,3-dicarlbonyl reactant in this reaction,anti-product P2 was the major product,with good diastereoselectivity(>95:5 dr;Fig.5a).DFT calculations were used to evaluate the generation of diastereoselectivity.As shown in Fig.5b,based on our mechanistic study,the diastereoselectivity of 1,5-annulation using 1,3-dicarlbonyl reactant R3 was determined by the energy difference between the two ring-closing transition states,28-ts-antiand 29-ts-syn.DFT calculation showed that the relative free energy of transition state 28-ts-anti,leading totransproduct P2,was 1.1 kcal/mol lower than that of transition state 29-ts-syn.Therefore,trans-product P2 was the major product,with a calculated 72:28 dr.This was in good agreement with the experimental observation that P2 was the major product with>95:5 dr.The higher relative free energy of transition states 29-ts-synwas attributed to steric repulsion in its geometry.To obtain a clear view of the energy difference between these two transition states,they were subjected to noncovalent interaction(NCI)analysis to determine steric repulsion.As shown in Fig.5c,observable steric repulsion between the phenyl group and ester group was labeled in the geometry information of transition state 29-ts-syn,leading to this transitions state having a higher relative free energy.Therefore,thetrans-product could be generated easilyviatransition state 28-ts
anti.
DFT calculations using the M06-L functional were used to study the mechanism of the Cu(I)-catalyzed[4+1]annulation of vinyl indoles and carbene precursors.As a result,a mixing mechanism involving sequential Cu-assisted indole C3-alkylation and direct 1,5-annulation was proposed and proved by our theoretical study.The reaction starts with carbenation of the Cu(I)species by a diazo compounds to afford a Cu-carbene complex,which can undergo intermolecular nucleophilic attack by the indole C3 position to afford alkylation with release of the Cu(I)catalyst.The generated zwitterionic intermediate then undergoes intramolecular annulation in the absence of a Cu species to yield the cyclopentaindole product.Carbenation of the Cu(I)species was found to be the ratedetermining step of the entire process.Interestingly,the competition between 1,3-and 1,5-annulation was considered,showing that 1,3-annulation leads to a three-membered-ring product as the kinetic product,which is easily generated,while the five-memberedring product was more stable.Therefore,five-membered cyclopentaindole will be observed as the major product of this reaction.This theoretical prediction is consistent with experimental observations.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
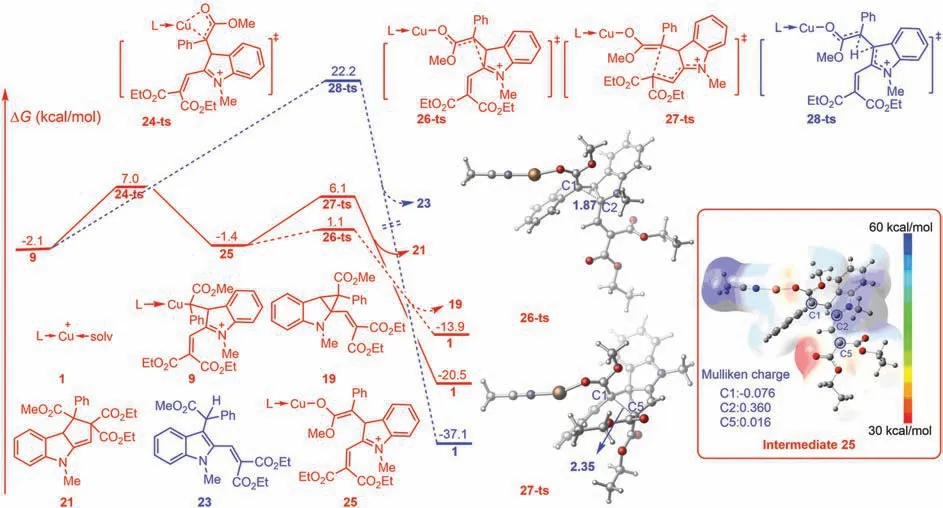
Fig.4.Gibbs energy profiles for the Cu-assisted annulation.The values given by kcal/mol are the relative free energies calculated by M06-L/6–311+G(d,p)/SMD//B3-LYP/6–31G(d)(SDD for Cu)method in dichloromethane solvent.Optimized geometries of transition states 26-ts and 27-ts.Bond lengths are shown in angstrom(˚A).Inset:Electrostatic potential map and Mulliken charge for intermediate 25.
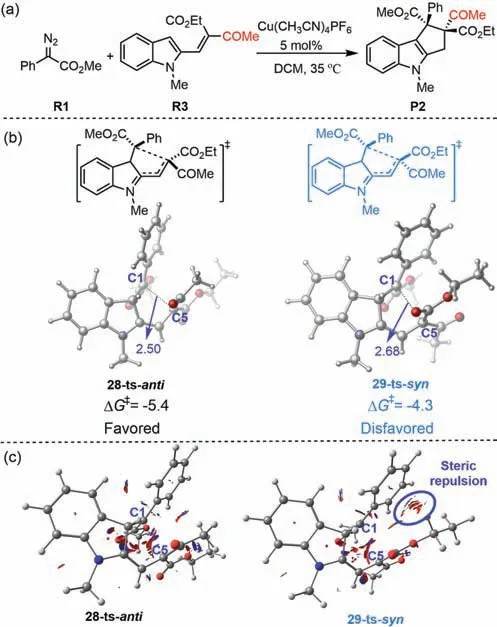
Fig.5.(a)Cu(I)-catalyzed[4+1]-annulation of asymmetric vinyl indoles and aryldiazoacetates.(b)Optimized structures of 28-ts-anti and 29-ts-syn(distances are given in angstroms(˚A)).The values given by kcal/mol are the relative free energies calculated by M06-L/6–311+G(d,p)/SMD//B3-LYP/6–31G(d)(SDD for Cu)method in dichloromethane solvent.(c)NCI plots for transition state 28-ts-anti and 29-ts-syn(blue indicates strong attraction,green indicates very weak interaction,and red indicates strong repulsion).
Acknowledgments
This work was supported by the National Natural Science Foundation of China(Nos.21822303,21772020 and 22003006).We are thankful for a project(No.2018CDXZ0002)supported by the Fundamental Research Funds for the Central Universities(Chongqing University).The project was supported by the Graduate Research and Innovation Foundation of Chongqing,China(No.CYB20045).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.10.010.
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis