
Electrochemistry enabled selective vicinal fluorosulfenylation and fluorosulfoxidation of alkenes
2022-06-20 06:21YiYuYiminJingShofenWuZhojingShiJinnnWuYofengYunKeyinYe
Chinese Chemical Letters 2022年4期
Yi Yu,Yimin Jing,Shofen Wu,Zhojing Shi,Jinnn Wu,Yofeng Yun,*,Keyin Ye,b,*
a Key Laboratory of Molecule Synthesis and Function Discovery(Fujian Province University),College of Chemistry,Fuzhou University,Fuzhou 350108,china
b State Key Laboratory of Physical Chemistry of Solid Surfaces,Xiamen University,Xiamen 361005,china
ABSTRACT Both sulfur and fluorine play important roles in organic synthesis,the life science,and materials science.The direct incorporation of these elements into organic scaffolds with precise control of the oxidation states of sulfur moieties is of great significance.Herein,we report the highly selective electrochemical vicinal fluorosulfenylation and fluorosulfoxidation reactions of alkenes,which were enabled by the unique ability of electrochemistry to dial in the potentials on demand.Preliminary mechanistic investigations revealed that the fluorosulfenylation reaction proceeded through a radical-polar crossover mechanism involving a key episulfonium ion intermediate.Subsequent electrochemical oxidation of fluorosulfides to fluorosulfoxides were readily achieved under a higher applied potential with the adventitious H2O in the reaction mixture.
Keywords:Alkene Chemoselectivity Electrochemistry Fluorine Sulfur
Alkenes are among the most prevalent and valuable feedstocks in organic synthesis.Direct and selective difunctionalization of alkenes,which simultaneously adds two synthetically valuable functionalities across a double bond,is a straightforward and economical approach to construct functionalized targets with high molecular complexities[1–3].Specifically,the direct incorporation of sulfur[4,5]and fluorine[6]into alkenes has attracted intensive research interest of synthetic chemists due to the important roles of both elements in organic synthesis,life science,and materials science.For instance,organic molecules containing both sulfur and fluorine are present in many well-received pharmaceuticals,such as Flovent,Faslodex,and Glecaprevir(Fig.1).Additionally,the spatial vicinity of these two elements further renders these types of compounds as ideal research targets of the fundamentally intriguing sulfur–fluorine gauche effect[7].It was found that the oxidation states of sulfur in these compounds are strongly correlated with their functions.Therefore,it is highly desirable to develop synthesis protocols to simultaneously introduce sulfur and fluorine elements into targeted molecules with precise control of the oxidation states of the sulfur moieties[8–10].
Typically,the vicinal fluorosulfenylation of alkenes is achieved by the reaction of alkenes with a chemically[11–17]or electrochemically[18]generated electrophilic thiolating agent(“RS+”)to form an episulfonium ion[19]followed by nucleophilic fluorination(Scheme 1A).Such a polar approach,however,is limited by the requisite preparation of highly reactive and toxic thiolating agents.Notably,Liu and co-workers[20]developed an elegant radical strategy for the intermolecular fluorosulfonylation of styrenesviaa high-valence palladium species[21,22].However,a superstoichiometric strong oxidizing agent(NFSI)was required(Scheme 1B).In contrast,the analogously direct fluorosulfoxidation of alkenes has not been reported yet due to the difficulty in accessing the electrophilic or radical sulfoxide species[23].
Green and sustainable electrosynthesis[24–31]could provide innovative solutions to address the challenges associated with conventional organic synthesis.To this end,one of the most prominent features of electrochemistry in organic synthesis is its unique capability to control reactivityvia"dialled-in" specific potential when necessary.By contrast,chemical agents only bear their innately fixed redox potentials and thus extensively screening of various chemical oxidants or reductants are generally required in a typical redox reaction.Therefore,electrosynthesis is capable to regulate reactions within a much wider redox window[32,33].In addition,the precise control of a minimally sufficient potential also allows better functional compatibility[34].In particular,electrochemical methods have been demonstrated to be capable of incorporating either sulfur[35,36]or fluorine[37–40]functionalities into diverse organic frameworks.Inspired by the elegant sulfonyl fluoride(RSO2-F)synthesisviathe electrochemical oxidative coupling of thiols(RSH)and potassium fluoride(KF)reported by Noël and co-workers[41,42],we speculated that electrochemistry should be an ideal solution to simultaneously[43]introduce both fluorine and sulfur in a controllable oxidation state into alkenes.Herein,we report an electrochemical radical-polar crossover approach for the highly selective fluorosulfenylation and fluorosulfoxidation of alkenes in which the selectivity was well controlled by the judicious choice of the applied potential(Scheme 1C).
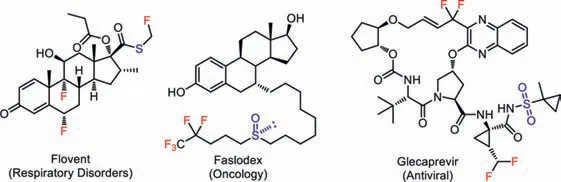
Fig.1.Pharmaceuticals containing sulfur and fluorine elements.
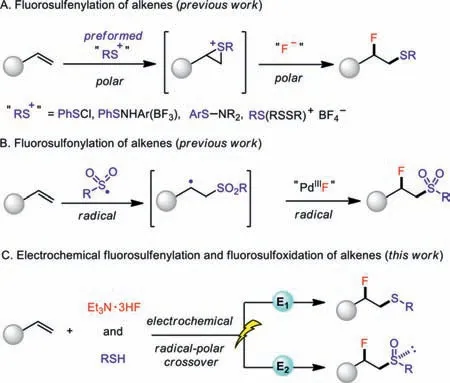
Scheme 1.Direct incorporation of sulfur and fluorine elements into alkenes.
We commenced our investigations on the direct vicinal fluorosulfenylation by choosing styrene(1),4-chlorothiophenol(2),and triethylamine trihydrofluoride(Et3N·3HF)as the model substrates.With a carbon cloth anode and a Pt cathode in an undivided cell,constant-cell-potential electrolysis(Ecell= 1.8 V)in MeCN at 40 °C delivered the desired fluorosulfide(3)in an optimal 77% isolated yield(Table 1,entries 1–3).No overoxidation of the sulfide product was observed under these conditions.Notably,Et3N·3HF in this reaction served not only as the fluorinating agent but also as the electrolyte owing to its ionic nature[44].The isolated yield of fluorosulfide was depressed along with the decrease of Et3N·3HF loading.Other fluorinating agents such as Olah’s reagent(Py·9HF),however,did not promote this transformation,and the formation of aryl disulfide was observed instead.A proof-of-concept experiment with the synthesis of fluorosulfoxide(4)by increasing the applied cell potential(2.5 V)was conducted(entry 4).Interestingly,the employment of CCl4(1 equiv.)as the additive was found to be pivotal for the formation of fluorosulfoxide[45,46].The higher applied cell potential(2.8 V)and the higher loading of styrene(1.7 equiv.)and Et3N·3HF(1 mL)were found to be beneficial(entries 5 and 6).Ultimately,the optimal yield of fluorosulfoxide(4,66%)was achieved under constant-current electrolysis(CCE)conditions(CCE at 20 mA for 4 h,entry 7).Note that the oxidation of fluorosulfide(3)to fluorosulfoxide(4)with a terminal chemical oxidant such asm-CPBA led to a low yield(47%)accompanied by the overoxidized fluorosulfone(see Supporting information for details).
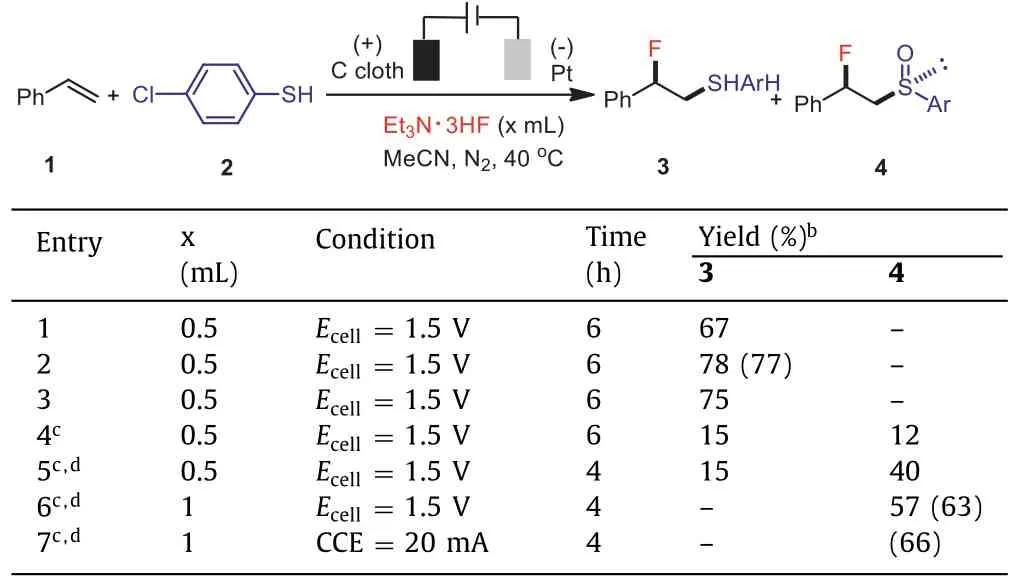
Table 1 Optimization of reaction conditions.a
With the optimal reaction conditions determined,we first evaluated the scope of the electrochemical fluorosulfenylation of alkenes(Scheme 2).This reaction accommodated a wide array ofpara-,meta-andortho-substituted styrenes(5–17).Additionally,alkyl(18–21),benzyl(22),pyridyl(23),and cycloalkyl(24–27)αsubstituted styrenes were all found to be well tolerated.Moreover,the gram-scale preparation of fluorosulfide 18(1.35 g,80%)further underscored the practicality of this protocol.Alkenes substituted by a naphthalene(28 and 29),alkene(30),alkyne(31),heterocycle(32–35)or estrone derivative(36)also underwent the desired transformations.This protocol was also readily transferred to the preparation of the analogous chlorosulfide(37)and bromosulfide(38).Furthermore,a variety of thiophenols bearing electrondonating or electron-withdrawing groups all reacted to afford the desired fluorosulfides in moderate to good yields(39–46,38%–87%).Encouragingly,cyclohexyl-(47),benzyl-(48)and heterocyclecontaining(49–52)thiols were all competent thiolating agents.Note that the applied cell potential was readjusted to 2.8 V when the electro-deficient 5-mercapto-1-methyltetrazole(51)and 2-mercapto-5-methyl-1,3,4-thiadiazole(52)were employed as the thiolating agents.
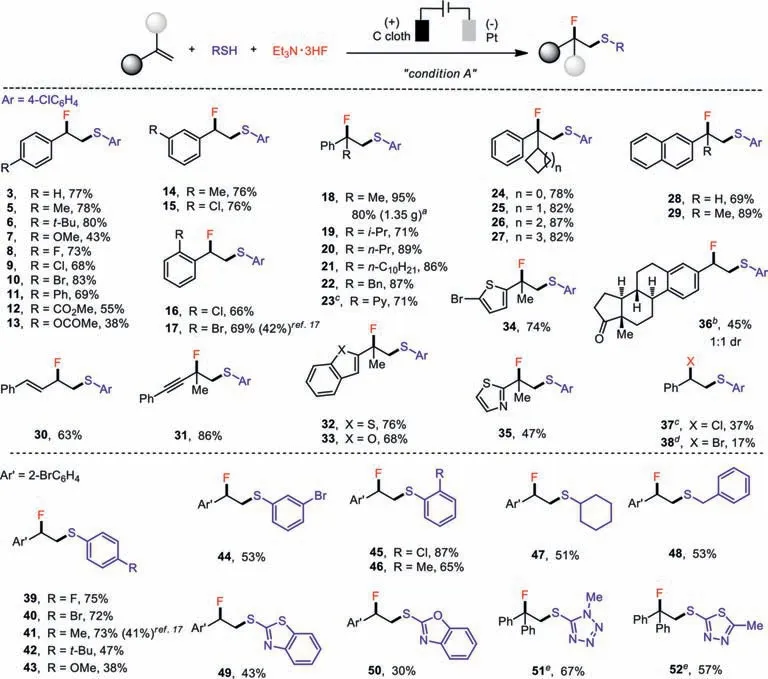
Scheme 2.Substrate scope of the electrochemical fluorosulfenylation of alkenes.Condition A:undivided cell,carbon cloth anode,Pt cathode,alkene(0.45 mmol),thiophenol(0.3 mmol),Et3N·3HF(0.5 mL),MeCN(10 mL)at 40 °C, Ecell = 1.8 V for 6 h,unless otherwise noted;yields of purified products; a gram-scale reaction,60 h; b 20 mA,75 min; c Et3N·HCl(5 equiv.)used instead; d Bu4NBr(3 equiv.)used instead; e 2.8 V,4 h.
Compared with the existing methods,this electrochemical fluorosulfenyaltion of alkenes exhibited several advantages.First,thiols were directly used to mitigate the tedious preparation of the highly reactive and toxic electrophilic thiolating agents as shown in Scheme 1A[11–18].Therefore,previously challenging alkyl and heterocycle substituted fluorosulfides could be readily obtained(Scheme 2).With respect to alkenes,literature protocols were typically restricted to electron-rich alkyl substituted ones.To the best of our knowledge,only fewortho-substituted styrenes were reported by Xu and co-workers in the fluorosulfenylation reaction usingN-thiosuccinimides in moderate yields(17 and 41)[17].In contrast,our method not only provided much higher yields of the same products but also tolerated a very broad scope of alkenes including styrenes,alkenyl,alkynyl,and heterocycle substituted alkenes(Scheme 2).Unfortunately,unactivated aliphatic alkenes were not well tolerated.Therefore,this electrochemical fluorosulfenyaltion should serve as a very general approach to fluorosulfides and is complementary to the existing methods.
We further applied this electrochemical difunctionalization to the facile preparation of synthetically challenging vicinal fluorosulfoxides directly from alkenes(Scheme 3).The target fluorosulfoxides were obtained as a pair of diastereoisomers since two stereogenic centers were generated.This reaction again was found to tolerate a diverse array of substrates,including substituted styrenes(53–62)and 1,1-diaryl alkenes(63–69).Our method was also well suited for the gram-scale preparation of the vicinal fluorosulfoxides 4(0.77 g,46% yield)and 63(1.19 g,56%yield).The structure of fluorosulfoxide 63 was unambiguously confirmed by X-ray diffraction analysis,which featured a profound sulfur-fluorine gauche effect(φFCCS(O)= 50.6°).Furthermore,1,1-disubstituted aryl ethylenes containing a methyl(70 and 71),cyano(72),cycloalkyl(73 and 74),heterocycle(75 and 76)group reacted to yield the desired fluorosulfoxides readily.Conjugated diene(77),enyne(78)and trisubstituted alkene(79)were also well tolerated.The vicinal fluorosulfoxidation of cyclic alkenes such as indene(80)and 1,2-dihydronaphthalene(81)afforded exclusivelytrans-difunctionalization products.With respect to the thiolating agents,a multitude of thiophenols(82–87)and alkyl(88–91),cyclohexyl(92),allyl(93)and benzyl(94)mercaptans were all able to provide various vicinal fluorosulfoxides in moderate to good yields(25%–71%).The relatively low yields of fluorosulfoxidation with aliphatic mercaptans was consistent with the observation of appreciate amounts of alkyl disulfides even after electrolysis.
To gain some insights into the reaction mechanism,several mechanistic experiments were then conducted.Replacement of the 4-chlorothiolphenol by its disulfide derivative(96)under standard conditions led to the desired fluorosulfide(3,67%)and fluorosulfoxide(63,51%)products(Scheme 4A).Consistent with this result,disulfide species were constantly observed during electrolysis,suggesting that thein-situ-generated disulfide might be a viable intermediate.The involvement of radical intermediates was substantiated by a radical rearrangement experiment(Scheme 4B)[47].Additionally,the stereospecific trans-fluorosulfenylation of indene(100)and 1,2-dihydronaphthalene(102)indicated that the reaction mechanism proceeded through an episulfonium ion intermediate(Scheme 4C).Interestingly,both the(Z)-and(E)-stilbenes(104)were transformed to fluorosulfide(105)with the same stereochemistry(Scheme 4D).Monitoring the reaction revealed that a facileZ→Eisomerization of(Z)-stilbene[48]occurred before the anticipated fluorosulfenylation,which was likely a thiyl-radicalmediated process[49].The oxygen atoms in the sulfoxide product likely originated from the adventitious H2O in the reaction mixture[50]rather than O2,as a similar yield of fluorosulfoxide(63)was obtained under rigorously oxygen-free conditions.This was further substantiated by the O18isotope labeling experiments,which showed that the degree of O18incorporation in the fluorosulfoxide(63)was roughly proportional to the amount of H2O18added(Scheme 4E)
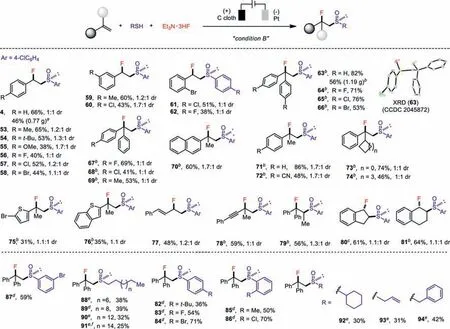
Scheme 3.Substrate scope of the electrochemical fluorosulfoxidation of alkenes.Condition B:undivided cell,carbon cloth anode,Pt cathode,alkene(0.51 mmol),thiophenol(0.3 mmol),Et3N·3HF(1 mL),CCl4(1 equiv.),MeCN(10 mL)at 40 °C,CCE at 20 mA for 4 h,unless otherwise noted;yields of purified products; a gram-scale reaction,CCE at 160 mA for 6 h; b 3.0 equiv.of CH3COOH were added; c 3.5 h; d 2.8 V; e CCE at 15 mA for 5 h; f 55 °C.
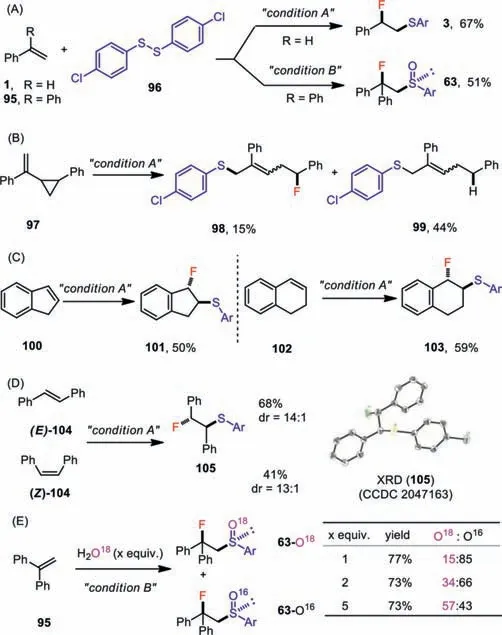
Scheme 4.Mechanistic experiments.
The highly selective fluorosufenylation and fluorosulfoxidation reactions well demonstrated the unique ability of the electrosynthesis to control the reactivityvia“dialled-in”potentials.Cyclic voltammetry studies further supported that a judicious choice of an applied potential(Ecell= 1.8 V,Eanode= 0.99–1.19 Vvs.Ag/AgCl during the electrolysis)was the key to achieving selective fluorosulfenylation without overoxidation(Fig.2,top).Additionally,sampling experiments of fluorosulfoxidation(62)showed that the first hour of electrolysis only led to the accumulation of fluorosulfide(39,Fig.2,bottom).Further oxidation to fluorosulfoxide(62)was observed thereafter along with the increase in the anodic potential(CCE at 20 mA,Eanode= 1.10–1.95 Vvs.Ag/AgCl during the electrolysis).
A proposed mechanism is shown in Scheme 5.Thiophenol(A)first underwent anodic oxidation to form a thiyl radical(B),which readily dimerized to disulfide(C)[51,52].This disulfide(C)was further oxidized anodically[53]to a thiyl radical(B)for subsequent addition to the alkene[54].An alternative route to this thiyl radicalviacathode reduction of disulfide(C)was possible[55]but not requisite in line with the success of this reaction even in a divided cell(see Supporting information for details).An episulfonium ion(E)could then be anticipatedviaan additional oxidation event(path a).However,an alternative pathway for forming this episulfonium ion from the reaction between the alkene and arylbis(arylthio)sulfonium ion(F)was also possible(path b)[56].At this stage,nucleophilic attack of the fluoride to the episulfonium ion formed a corresponding fluorosulfide(G)[57].The oxidation state of sulfur can be further fine-tuned by applying a higher cell potential to generate fluorosulfoxide(H).Though the exact roles of CCl4in this electrochemical oxidation of fluorosulfide to fluorosulfoxide still need to investigate in detail,the reductive generated chlorospecies(Cl—or Cl·)from CCl4was proposed to facilitate this oxidation process[58–59].Further attempts to access the fluorosulfone(I)with increases of the applied potential(Ecell= 2.8–3.8 V),however,only led to the decomposition of the starting material.
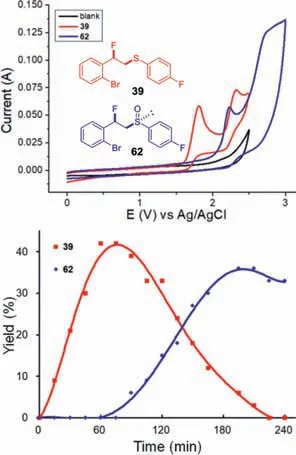
Fig.2.Top:Cyclic voltammograms of fluorosulfide(39)and fluorosulfoxide(62).Bottom:Sampling experiments of fluorosulfoxidation(62,CCE = 20 mA,condition B).See Supporting information for details.
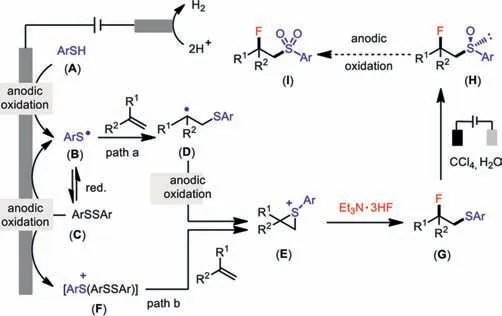
Scheme 5.Proposed mechanism of the electrochemical fluorosufenylation and fluorosulfoxidation.
In conclusion,we developed a highly selective,appliedpotential-controlled process for the vicinal fluorosulfenylation and fluorosulfoxidation of alkenes.The protocol allowed the facile preparation of a diverse array of fluorosulfides and fluorosulfoxides that are otherwise challenging to obtain.Mechanistic investigations revealed that the judicious choice of an applied potential is the key to achieving high selectivity.Such a unique feature of electrosynthesis to control the reactivityvia“dialled-in”potentials could serve as a conceptional inspiration for other new reaction designs.We speculated that this protocol will find broad applications for the synthesis of sulfur-and fluorine-containing molecules in the life science and materials science.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Financial support from the National Natural Science Foundation of China(Nos.21901041 and 21772023),State Key Laboratory of Physical Chemistry of Solid Surfaces,Xiamen University(No.202008),and Fuzhou University(No.510841)is gratefully acknowledged.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.10.016.
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis