
Cucurbit[7]uril-threaded flexible organic frameworks:Quantitative polycatenation through dynamic covalent chemistry
2022-06-20 06:20QinLiJinSunBoYngHuiWngDnWeiZhngZhnTingLi
Chinese Chemical Letters 2022年4期
Qin Li,Jin-D Sun,Bo Yng,Hui Wng,Dn-Wei Zhng,*,D M,Zhn-Ting Li,*
a Department of Chemistry,Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials,Fudan University,Shanghai 200438,China
b College of Chemistry,Zhengzhou University,Zhengzhou 450001,China
ABSTRACT A three-dimensional flexible organic framework FOF-1 has been synthesized from the condensation of a tetratopic acylhydrazine and a rigid 4,4′-diphenyl-4,4′-bipyridinium dialdehyde in water through the quantitative formation of hydrazone bond.FOF-1 is further applied to construct a polycatenane framework FOF-pc-1 through the quantitative cucurbit[7]uril encapsulation for the diphenylbipyridinium subunits of the framework by making use of the dynamic nature of the hydrazone bond in water.The bipyridinium subunits in both frameworks can be reduced their radical cation counterparts to produce conjugated radical cation-linked dynamic organic frameworks rc-FOF-1 or rc-FOF-pc-1.Polycatenation is revealed to enhance the stability of the dynamic frameworks in water,whereas depolycatenation can be reached for both FOF-pc-1 and rc-FOF-pc-1 by using a ferrocene guest to form a more stable complex with CB[7].
Keywords:Polycatenation Flexible organic framework Dynamic covalent chemistry Cucurbit[7]uril Bipyridinium Radical cation
Mechanically interlocked molecules,including rotaxanes,catenanes and knots,have been demonstrated to be versatile for the construction of topologically interesting structures and molecular machines[1–5].Polycatenanes are supramolecular polymers that are composed entirely of interlocked rings extending in one-,two,or three-dimensional space[6,7].Such kind of mechanically interlocked polymers represent useful platforms for exploiting new properties or functions that are not obtainable from conventional covalent polymers.One of the prominent examples is the so-called slide-ring gels ofα-cyclodextrin-derived supramolecular polymers,where the mechanically interlocked cyclodextrin rings act as mobile cross-linking sites of coatings for cell phones and automobiles to exhibit remarkable anti-scratch and healing characteristics[8].However,the construction of polycatenanes has been a major challenge.Most of reported works on polycatenanes has focused on linear poly([2]catenane)s that are formed by polymerizing discrete bisfunctionalized[2]catenanes or efficient ring closing of metallosupramolecular polymers[9–13].Loeb has utilized 24-crown-8-based[2]pseudorotaxanes to construct two-and three-dimensional(3D)metal–organic rotaxane frameworks using stable coordination complexes as the connecting nodes[14,15].These structurally defined architectures contain mechanically interlocked units,but can only maintain the interlocking in the solid state.Thus,there is a high demand for the construction of new kinds of polycatenanes for exploring interlocking-derived unique properties of polymeric structures.
In the past two decades,dynamic covalent chemistry has been widely used for the preparation of dynamic covalent polymers[16–25].Many of processible covalent organic polymers have been investigated as advanced adaptive materials that exhibit functions such as self-healing,shape memory,recyclability,degradability or stimuli responsiveness[26–31].In an effort to achieve watersoluble polymers with intrinsic nanoscale porosity,we recently reported the construction of flexible organic frameworks(FOFs)from tetratopic molecular components through the quantitative formation of the hydrazone bond in water[32–34].As a new class of dynamic porous organic polymers that are highly soluble in water,FOFs have been applied as nanoscale carriers for encapsulating proteins and DNA for intracellular delivery and as frameworks for the design of prodrugs for enhancing the antitumor effi-cacy of anthracycline drugs.Herein we describe the self-assembly of a flexible organic framework polycatenane FOF-pc-1 in water through the quantitative threading of cucurbit[7]uril(CB[7])onto the 4,4′-diphenyl-4,4′-bipyridinium units that are incorporated in a 3D flexible organic framework FOF-1.We demonstrate that polycatenation significantly improves the stability of the framework and 4,4′-bipyridinium(BIPY2+)units in both FOF-1 and FOF-pc-1.The BIPY2+units of both FOFs can be quantitatively reduced to BIPY·+radical cations,leading to the formation of conjugated radical cations-linked rc-FOF-1 or rc-FOF-pc-1.Moreover,the CB[7]rings of FOF-pc-1 can efficiently inhibit the dimerization of the BIPY·+units of the framework,an intrinsic feature of conjugated radical cation species[35–38],by forming more stable 1:1 complexes[39,40].
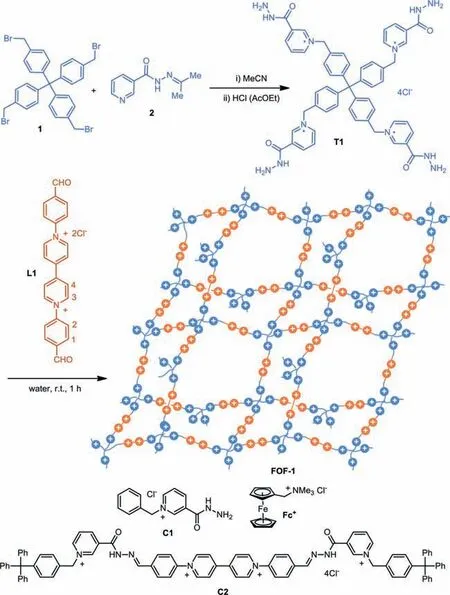
Fig.1.Synthesis of hydrazone-based cross-linking 3D FOF-1 and the structures of control compounds C1,Fc+ and C2.
Previous studies have illustrated that multitopic aldehyde and acylhydrazine components can condense in water to afford hydrazone-connected FOFs quantitatively[32–34].Compounds T1 and L1 were used to construct BIPY-incorporated flexible organic frameworks(Fig.1).T1 was prepared from the reaction of compounds 1 and 2,which was followed by treatment of hydrogen chloride,whereas L1 has been used to prepare hydrazone-based linear dynamic covalent polymers[41].1H NMR in D2O showed that T1(1.2 mmol/L)and L1(molar ratio = 1:2)readily condensed to afford hydrazone bonds quantitatively at room temperature,which was confirmed by the observation that the diagnostic O=CH signal of L1 at 10.1 ppm vanished completely after about 1 h(Fig.2a).Given the sensitivity of the1H NMR method,we might reasonably assume that the hydrazone bonding was formed in ≥97% yield.The CHO groups of L1 were partially hydrated to afford CH(OH)2,CH(OD)2in D2O,in about 10% yield.The CH signal appeared at 6.25 ppm,which also disappeared in the1H NMR spectrum of the mixture.The resolution of the spectrum decreased considerably,suggesting the formation of new hydrazonebased flexible organic framework FOF-1.Increasing the temperature to 50 °C caused the occurrence of the weak CHO peak in the spectrum,and the peak was enhanced slightly at 90 °C,indicating partial hydrolysis of the hydrazone bonding(Fig.2b).Upon cooling to room temperature,the signal vanished again,suggesting that the aldehyde converted to hydrazone again.Diluting the solution to[T1]= 0.8 mmol/L also caused the CHO signal to occur(Fig.S1 in Supporting informaiton),which showed that the hydrazone bond was partially hydrolyzed in the diluted solution.1H NMR experiments also revealed that,after reaching the equilibrium,the reaction of the control 1-benzylpyridin-1-ium-3-carbohydrazide C1(4.8 mmol/L)and L1(2.4 mmol/L)gave rise to the corresponding uni-and bi-hydrazone derivatives P1 and P2 with totally 85% conversion of the aldehyde to the hydrazone(Fig.S2 in Supporting information).Thus,quantitative formation of the hydrazone bonding from T1 and L1 clearly reflected a positive multivalency effect,through which the hydrazone bonds stabilized each other to lead to the generation of FOF-1(Fig.2).The infrared spectrum of FOF-1 showed that the stretching vibration of the aldehyde C=O bond of L1,centered at 1703 cm-1,disappeared completely(Fig.S3 in Supporting informaiton),which also supported the quantitative formation of the hydrazone bonding.
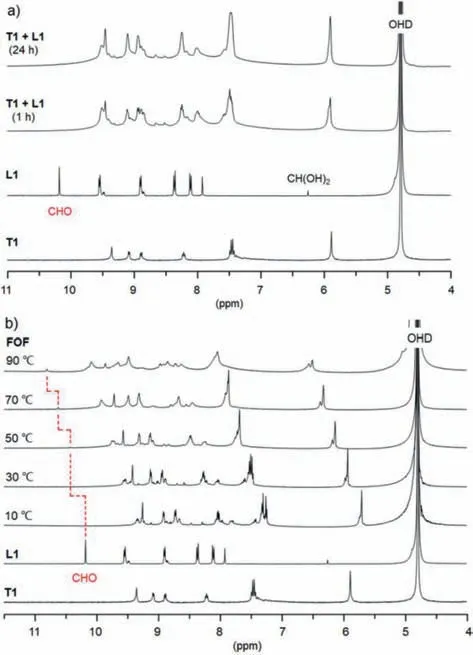
Fig.2.(a)1H NMR spectrum(400 MHz)of T1(1.2 mmol/L),and L1(2.4 mmol/L)and FOF-1(T1+L1,1:2)with different reaction time in D2O(pD = 6.5)and(b)1H NMR spectrum(500 MHz)of T1,L1 and FOF-1([T1]= 1.2 mmol/L)in D2O at different temperature.
Dynamic light scattering(DLS)experiments revealed that FOF-1 existed as a nanoscale polymer.Within the concentration range of[T1]= 0.06 mmol/L to 1.0 mmol/L,the aqueous solutions gave rise to a hydrodynamic diameter(DH)of 38 nm to 91 nm(Fig.3a).Similar values were obtained after the solutions were left to stand for 3 months,which supported the high stability of the polymer.The solution of T1 at 1.2 mmol/L afforded a much smaller DHof about 10 nm(Fig.3a),which suggested that T1 itself could underwent important intermolecular stacking.However,DLS did not revealed the formation of nanoscale entities by L1 even at a high concentration(4.8 mmol/L).The above1H NMR experiments indicated that the hydrazone bond of FOF-1 was partially hydrolyzed at a low concentration([T1]=<0.8 mmol/L)(Fig.S1).Thus,the above DLS results supported that the unhydrolyzed hydrazone linkers could still connect the two components to afford nanoscale polymer framework.At[T1]= 1.2 mmol/L,the zeta potential of FOF-1 was determined to be 53.2 mV,which indicated that the framework possessed a positively charged surface as a result of the cationic character of the tetrahedral component.Previous guest adsorption experiments confirmed that this family of flexible organic frameworks possess inherent pores in solution[32,33].Molecular modeling study revealed that the pore aperture of the macrocycle formed through the 6+6 condensation of the two components that adopted extended conformations was about approximately 73.9 nm.
Compound L1 has four aromatic subunits.Crystal structure analysis revealed that L1 formed 1:2 complex with CB[7],where two CB[7]rings encapsulated the two benzene units(Fig.3c),which was consistent with the reported complexation between CB[7]and 4,4′-diarylbipyridiniums that depends on the substituents on the two aryl rings of the latter[42–47].1H NMR of their 1:2 mixture in D2O([L1]= 1.0 mmol/L)also supported this 1:2 encapsulation pattern(Fig.S4 in Supporting information),as the H-1,H-2 and H-3 signals(Scheme 1 for numbering)and the O=CH signal of L1 all underwent significant upfield shifting(Δδ0.82,0.94 and 0.34 ppm),which may be attributed to the shielding effect of the CB[7]ring through encapsulation.In contrast,the H-4 signal of the pyridine subunits suffered notable downfield shifting(Δδ0.19 ppm),which should reflect the deshielding effect of the CB[7]ring.By using isothermal calorimetric titrations(Fig.S5 in Supporting information),the apparent association constant(Ka)of the 1:1 complex between CB[7]and the benzene ring of L1 was determined to be 7.3(±1.9)× 105L/mol.The relatedΔHand-TΔSwere-8.6(±0.15)and 0.61 kcal/mol,respectively,indicating that the complexation was driven enthalpically.
Since CB[7]has been demonstrated as a robust rigid macrocyclic host for selective encapsulation of discrete aromatic units in water[48–53],its encapsulation for the different subunits of FOF-1 was then studied.Addition of CB[7]caused important change of the signals of FOF-1 in1H NMR spectrum in D2O(Fig.S6 in Supporting information),suggesting the encapsulation of the BIPY2+subunits by the macrocycle molecules.This encapsulation should take place through the hydrolysis of the hydrazone bonds followed by re-formation.We also found that the above1H NMR spectrum was time-independent,indicating that the encapsulation took place very quickly on the1H NMR time scale.However,the1H NMR spectrum had a quite low resolution and thus did not provide useful information for the binding stoichiometry between CB[7]and the different subunits of FOF-1.FOF-1 exhibited a strong absorption band centered at 385 nm(Fig.S7 in Supporting information).We thus recorded the influence of the addition of CB[7]to this absorption band.It was found that adding CB[7]caused important hypochromic effect for this absorption band,but an inflection point was observed after the addition of 2 equiv.of CB[7],which was relative to[L1].This observation supported that the L1 subunits of FOF-1 were also encapsulated by CB[7]with a 1:2 stoichiometry to afford the CB[7]-BIPY2+-interlocking flexible organic framework FOF-pc-1.To gain more evidence for this binding stoichiometry,control compound C2 was further prepared.Its absorption band around 265 nm exhibited similar hypochromic effect(Fig.S8 in Supporting information),which also reached maximum with the addition of 2 equiv.of CB[7],further confirming the formation of the FOF polycatenane.Job plot experiments were also carried out using the absorption method(Fig.S9 in Supporting information),which again supported the 1:2 encapsulation stoichiometry.Given the dynamic feature of the hydrazone binding,it is reasonable to propose that there might be a small amount of free CB[7]rings existing in the solution,which reached equilibrium with the interpenetration-engaged ones.
At 50 °C,the1H NMR spectrum of FOF-pc-1 did not display observable peak of the CHO group,which began to appear only at 90 °C(Fig.S10 in Supporting information).Thus,the catenation of the BIPY2+subunits by CB[7]actually considerably enhance the stability of the framework.DLS experiments showed that CB[7]polycatenation caused slight increase of the DHof the framework of different concentrations(Fig.3b).For example,at[T1]= 0.06 and 1.0 mmol/L,the DHvalue of the framework was increased from 38 and 91 nm to 44 and 106 nm,respectively,which may be rationalized by considering that catenation produced steric hindrance to force the linkers to adopt more extended conformations.As a result of polycatenation,FOF-pc-1 also exhibited a reduced zeta potential(32.4 mV),indicating that the CB[7]imposes a shielding effect to lead to the reduction of the positive potential of the framework surface.
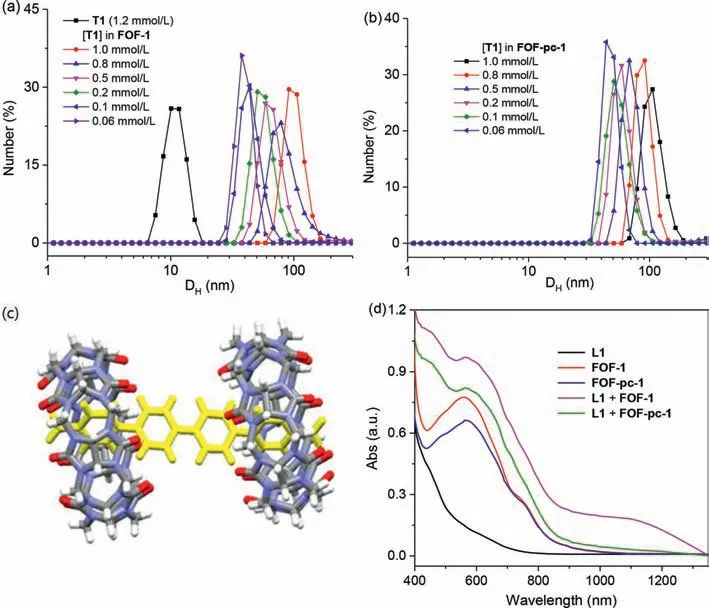
Fig.3.(a,b)DLS profiles of T1 and FOF-1 and FOF-pc-1 of different concentrations,represented by[T1],in water at 25 °C.(c)Crystal structure of the L1·2CB[7]complex(CCDC:2,075,839).(d)UV–vis spectrum of L1(0.1 mmol/L),FOF-1,FOF-pc-1,L1+FOF-1 and L1+FOF-pc-1 in H2O in the presence of sodium dithionite(50 mmol/L)at 25 °C.[BIPY]= 0.1 mmol/L for all the solutions.
It is reported that cationic ferrocene derivative Fc+(Fig.1)forms a highly stable 1:1 complex with CB[7]in water,with aKaof 4 × 1012L/mol[54].1H NMR spectrum in D2O showed that adding 1.0 equiv.of Fc+,which was relative to CB[7],to the solution of FOF-pc-1 led to exclusive decatenation of the framework to re-form FOF-1,because Fc+was completely complexed by CB[7]to exhibit a new set of peaks(Fig.S11 in Supporting information),which could be realized only through the decatenation of the framework to release the CB[7]molecules.
The BIPY2+unit can be readily reduced by sodium dithionite in water to radical cation BIPY·+[55].The UV–vis absorption spectra of both FOF-1 and FOF-pc-1 were thus recorded in water in the presence of sodium dithionite(Fig.3d).As expected,both solutions exhibited a strong absorption band centered at 566 and 556 nm[55],respectively,which corresponded to that of their BIPY·+unit and supported the formation of new radical cation-linked frameworks rc-FOF-1 and rc-FOF-pc-1(Fig.4).However,for both solutions,no absorption band of the(BIPY·+)2dimer was observed,which typically appeared within the range of 800-1300 nm[55].Clearly,the formation of the framework prevented the stacking of the BIPY·+units.The spectrum of L1 gave rise to a very weak absorption band around 600 nm(Fig.3d),suggesting that it underwent similar one-electron reduction.Electron paramagnetic resonance spectrum of the three solutions all displayed a broad signal of comparable intensity(Fig.S12 in Supporting information)[44],which also supported the formation of the radical cations.DLS experiments revealed that rc-FOF-1 and rc-FOF-pc-1 at different concentrations gave rise to DHthat were comparable with that of FOF-1 or FOF-pc-1 of the same concentration(Fig.S13 in Supporting information),verifying that the frameworks were still stable in the radical cation state.UV–vis titration experiments indicated that adding Fc+to the solution of rc-FOF-pc-1 could lead to the re-formation of FOF-pc-1 through decatenation to release CB[7]to form more stable complex CB[7]⊂Fc+(Fig.S14 in Supporting information).
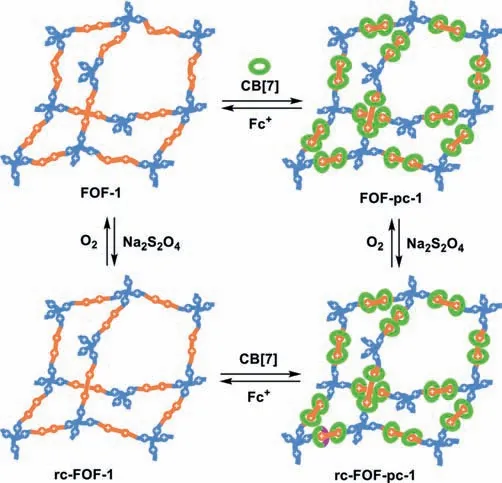
Fig.4.Schematic presentation of BIPY-linked flexible organic frameworks and their recyclable transformation.
Adding L1 to the solution of rc-FOF-1 led to the appearance of an absorption band centered at 1100 nm,which was the characteristic absorption of the(BIPY·+)2dimers.Because the respective solutions did not give rise to similar absorption,this dimerization should mainly take place between the BIPY·+unit of L1 and that of rc-FOF-1.This result also suggested that the molecules of L1 might be enriched in the pores of rc-FOF-1.In contrast,no similar absorption was observed from the mixture solution of L1 and rc-FOF-pc-1,indicating that the CB[7]macrocycles prevented the threaded BIPY·+units from similar stacking.UV–vis absorption spectroscopy further showed that,upon exposing the solution of rc-FOF-1 and rc-FOF-pc-1 to air,their BIPY·+units were rapidly oxidized to BIPY2+lead to the recovery of FOF-1 and FOF-pc-1(Fig.4).
We have demonstrated that polycatenated 3D flexible organic frameworks can be synthesized by combining dynamic covalent chemistry and strong encapsulation of CB[7]for hydrophobic 4,4′-diphenyl-4,4′-bipyridinium in water.The original dynamic covalent framework and the more complicated polycatenated framework can be further transformed to their radical cation-featured counterparts.Polycatenation considerably enhances the stability of the polycationic dynamic organic frameworks in water.Polycatenation can also inhibit the inherent dimerization of conjugated radical cations,which may be useful for exploiting new properties of unstacking conjugated radical cation species.Notably,CB[7]rings can be removed from the frameworks by adding a ferrocene guest that forms more stable encapsulation complexes with CB[7].The recyclability of the different frameworks well illustrates the robustness of dynamic covalent chemistry in constructing advanced hierarchical macromolecular systems.In the future,this new polycatenation strategy will be explored for the construction of neutral and processible interlocked architectures that may exhibit new elastomeric property.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The work was financially supported by the National Natural Science Foundation of China(Nos.21890732,21890730 and 21921003).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.10.017.
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis