
Triphenylamines consisting of bulky 3,5-di–tert–butyl–4-anisyl group:Synthesis,redox properties and their radical cation species
2022-06-20 06:19ManfeiZhouLijunMaoYanFeiNiuXiaoLiZhaoXueliangShiHaiBoYang
Chinese Chemical Letters 2022年4期
Manfei Zhou,Lijun Mao,Yan-Fei Niu,Xiao-Li Zhao,Xueliang Shi,Hai-Bo Yang
Shanghai Key Laboratory of Green Chemistry and Chemical Processes,School of Chemistry and Molecular Engineering,East China Normal University,Shanghai 200062,China
ABSTRACT Triphenylamine(TPA)derivatives have been widely used as useful building blocks for diverse functional materials because of their excellent redox activity.Most of the molecular structures of TPA-based organic functional materials contain 4-anisyl groups,which on one hand could reduce their oxidation potential and on the other hand significantly delocalize the spin density of the resultant TPA radical cation species and enhance their stability.However,molecular-level investigation of the redox behavior of triphenylamines consisting of 4-anisyl group and the electronic structures of their radical cation species has not been reported in the literature.Herein,we design a series of triphenylamines consisting of one,two,or three 3,5-di–tert–butyl–4-anisyl groups and investigate their redox behaviors and corresponding radical cation species.We disclose that the resonance hybrid and steric protection could both contribute to the stability of triphenylamine radical cations.Moreover,further oxidation leads to an unexpected oxidative demethylation.The findings in this work may reveal new insights for the understanding of the unique redox properties of 4-anisyl substituted triphenylamines.
Keywords:Triphenylamine Radical cation Stability Redox Steric protection
Triphenylamine(TPA)together with its derivatives and their radical cation counterparts are well known for their excellent optical[1–3],electric[4–5],and magnetic properties[6–9],which renders TPA core as one of the most extensively utilized building blocks in many organic functional materials[10–22].For example,TPA derivatives have been widely employed as the hole-transport layers(HTLs)for dye sensitized solar cells(DSSCs)and perovskite solar cells(PSCs)because of their excellent redox activity[23–26].Most of the molecular structures of TPA-based HTLs contain 4-anisyl group,which on one hand could reduce their oxidation potential and facilitate the hole injection process,and on the other hand significantly delocalize the spin density of the resultant TPA radical cation species and enhance their stability[27–30].Therefore,the investigation of the redox behavior of TPA consisting of 4-anisyl group is of great interest.
Triphenylamine-based radical cation is reasonable persistent when thepara-position of TPA core is properly protected[31–45].Thepara–methoxy substituted TPA radical cations in principle exhibit enhanced persistence because such species could be represented by a resonance hybrid wherein the spin density is overall delocalized in the 4-anisyl group(Scheme 1)[46].Besides,the dication of thepara–methoxy substituted TPA was also speculated[47].However,such resonance hybrid has rarely been directly confirmed experimentally,especially in characterizing electronic structures through X-ray crystallographic analysis.Moreover,thepara–methoxy substituted TPA radical cations might still suffer significant issue in chemical instability if theortho-position is not adequately protected.Recently,we demonstrated that 3,5-di–tert–butyl–4-methoxyphenyl group at the nitrogen atom of the carbazole could effectively reduce the ionization energy of carbazole and enhance the stability of its radical cation[48].In this scenario,we anticipate that the bulky 3,5-di–tert–butyl–4-anisyl group may have a profound effect on the stability of TPA radical cation species because of the additional steric protection,which meanwhile would give a detailed insight into the resonance structures.
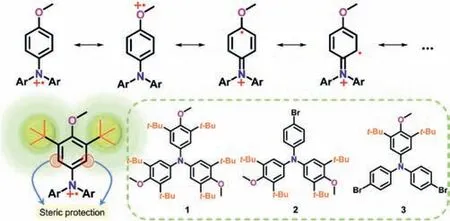
Scheme 1.The major resonance structures of TPA radical cation consisting of 4-anisyl group,the proposed steric protecting strategy and the target molecules in this work.
Herein we design and synthesize a series of TPA molecules(1,2 and 3)consisting of one,two,or three 3,5-di–tert–butyl–4-anisyl groups(Scheme 1),and systematically investigate the substitution effect on their redox behavior and the stability of the resultant radical cations.Cyclic voltammetry experiment demonstrated that the introduction of anisyl groups significantly reduced the oxidation potential of TPA molecules and simultaneously enhanced the stability of the TPA radical cations.Consequently,the radical cation species of 1·+-3·+were successfully obtained and their structures were unambiguously determined by X-ray crystallographic analysis.The photophysical properties of 1·+-3·+and their electronic structures were systematically investigated by UV–vis-NIR spectroscopy and electron paramagnetic resonance spectroscopy(EPR),assisted by density functional theory(DFT)calculations.The results implied that the resonance hybrid and steric protection could both contribute to the stability of triphenylamine radical cations 1·+-3·+.Moreover,an unexpected oxidative demethylation was observed when 1 was further oxidized by a stronger oxidant,resulting in a quinone like structure of 1-Q as confirmed by X-ray crystallographic analysis.We anticipate the findings in this work will shed some light on the design of novel triphenylamine-based radical cations and related materials.
As shown in Scheme 1,a series of TPA molecules(1,2 and 3)consisting of one,two,or three 3,5-di–tert–butyl–4-anisyl groups were designed and synthesized.Compound 1 was synthesized through a facile one-step Pd-catalyzed amination reaction[49]from 1–bromo-3,5-di–tert–butyl–4-methoxybenzene and urea which is serving as ammonia equivalent in moderate yield(75%)(Experimental section and Scheme S1 in Supporting information).Compounds 2 and 3 were prepared through Buchwald-Hartwig amination of 1–bromo-3,5-di–tert–butyl–4-methoxybenzene with phenylamine followed by the bromination reaction withNbromosuccinimide.The structures of compound 1–3 were thoroughly characterized by1H and13C NMR,HR-MS measurements as well as single crystal X-ray crystallography(Supporting information).
The redox properties of triphenylamines 1–3 were then evaluated by cyclic voltammetry(CV)in CH2Cl2at 298 K with 0.1 mol/L tetrabutylammonium hexafluorophosphate as supporting electrolyte(Fig.1a).Triphenylamines 1–3 all exhibited two oxidation waves,and the half-wave potentials(Eox1/2,potentials are referredvs.Fc/Fc+)of 1–3 were determined to be+0.01 V,+0.20 V,and+0.41 V,respectively.No reduction wave was observed for 1–3.The oxidation potentials significantly decreased in an order from 3,2 to 1,indicating that the 3,5-di–tert–butyl–4-anisyl group could remarkably reduce the oxidation potential of TPA core.Notably,in all cases,the first oxidation wave was reversible while the second one was irreversible,implying the formation of radical cation was feasible but might be difficult in achieving the dication species(vide infra).
Motivated by the results of cyclic voltammetry,we tried to synthesize the radical cation species and investigate their electronic structures,and compare the difference between each radical cation species,as well as between the neutral triphenylamines and triphenylamine radical cations.The radical cation species of 1·+-3·+were obtained by single electron oxidation of 1–3 in the presence of AgSbF6in nearly quantitative yield(Scheme S2 in Supporting information).The UV–vis-NIR spectra of 1–3 and 1·+-3·+were surveyed and compared.The UV–vis-NIR absorption profiles of 1–3 in CH2Cl2were nearly identical,all showing intense absorption in the 260–350 nm range with the maxima at 300,299 and 302 nm for 1,2 and 3,respectively(Fig.1b,dash line).The almost same absorption onset of 1–3 implied that their energy gaps remained unchanged in spite of their different oxidation potential.The density functional theory(DFT)calculations revealed the substitution of 3,5-di–tert–butyl–4-anisyl groups obviously altered the HOMO/LUMO alignment of TPA core but without changing their energy gaps(Fig.S7 in Supporting information),which was consistent with the above CV and UV–vis-NIR analysis.The radical cation species 1·+-3·+exhibited obvious absorption in the visible and near-infrared region compared with the absorption of their neutral molecules(Fig.1b,solid line).The observed long wavelength absorption bands could be assigned to the HOMO →SOMO transitions based on the time-dependent density functional theory(TDDFT)(Tables S1-S3 in Supporting information).
The EPR spectra of radical cation species 1·+-3·+all exhibited three main peaks due to the nitrogen hyperfine splitting(mI=0,±1)with agtensor around 2.004,indicating the efficient spin delocalization on the N atom(Fig.2a,solid line).Each of the three peaks in 1·+further split into seven peaks due to the six equivalentortho-protons.Because of the existence of multiple inequivalentortho-protons,hyperfine structures of 2·+and 3·+were not fully resolved.The EPR spectra of 1·+-3·+can be roughly simulated through ORCA program at the UB3LYP/TZVP level of theory(Figs.S10-S12 in Supporting information).The spin density maps of 1·+-3·+were also calculated at the UB3LYP/6–311G(d,p)level of theory,and the spin density distribution disclosed some interesting information(Fig.2b and Fig.S7 in Supporting information).The results revealed that the spin densities were considerably delocalized on the anisyl groups in 1·+-3·+,even on the oxygen atom.Moreover,the spin densities on the anisyl groups were more prominent than those on thepara-bromophenyl groups,implying the main resonance structures in Scheme 1 were valid.
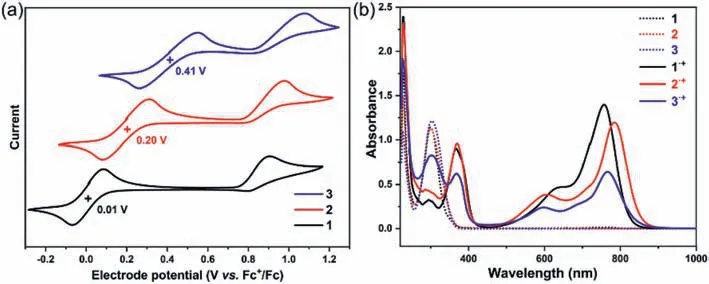
Fig.1.(a)Cyclic voltammograms of 1–3 measured in CH2Cl2(at 1.0× 10-3 mol/L)containing 0.1 mol/L n-Bu4NPF6 at 298 K(scan rate:20 mV/s).(b)UV–vis-NIR spectra of 1–3(dash line)and radical cations 1·+-3·+(solid line).
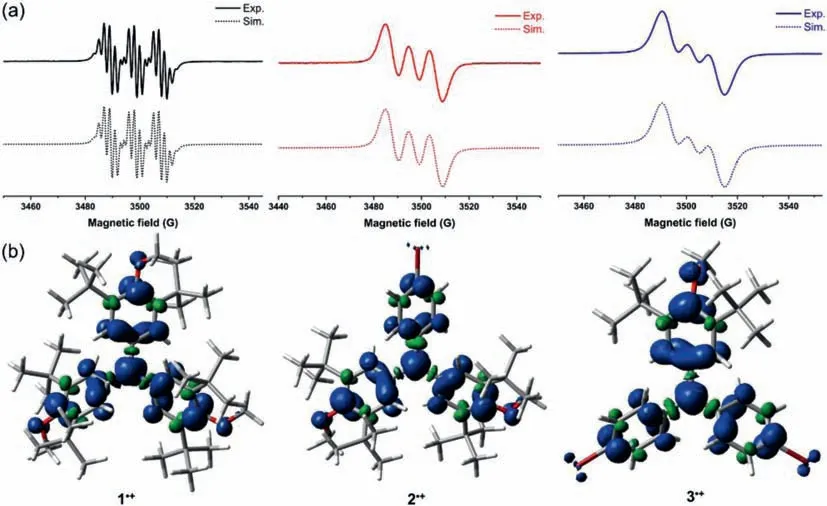
Fig.2.(a)EPR spectra and the simulations via EPR simulation programs of 1·+-3·+ in CH2Cl2 solution.(b)Spin density distributions of 1·+-3·+ at the UB3LYP/6–311G(d,p)level of theory.
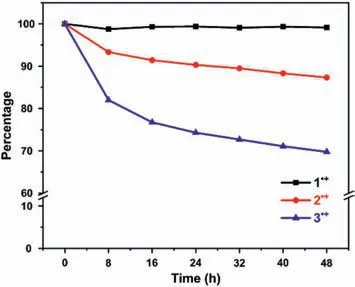
Fig.3.Evaluation of the stability of radical cation species of 1·+(black line),2·+(red line),and 3·+(blue line) via UV–vis-NIR spectroscopy.
In order to investigated the stability of the three triphenylamine radical cations 1·+-3·+,we measured the time-dependent absorption spectra of 1·+-3·+in CH2Cl2(Fig.3).Radical cations 1·+with three 3,5-di–tert–butyl–4-anisyl was found to be very stable after 48 h in ambient environmental conditions because its absorbance in CH2Cl2solution remained almost constant(Fig.3 and Fig.S1 in Supporting information).In contrast,radical cations 2·+and 3·+exhibited obvious decrease in their absorbances in CH2Cl2,especially for 3·+that was found to remain only 70% after 48 h(Fig.3,Figs.S2 and S3 in Supporting information).Therefore,the introduction of more bulky 3,5-di–tert–butyl–4-anisyl groups could significant enhance the stability of the TPA radical cations,mainly due to the effectively kinetic protection,which is also consistent with our recent study[48].
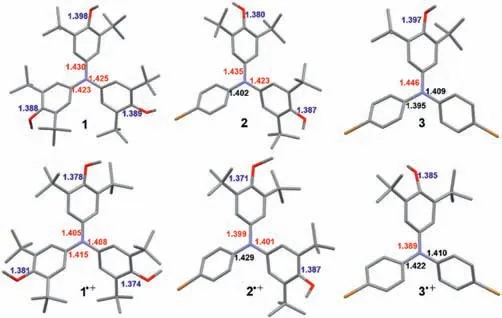
Fig.4.X-ray crystallographic structures of 1–3 and 1·+-3·+ and their selective bond lengths.Hydrogen atoms and SbF6- anions are omitted for clarity.
The structures of 1–3 and 1·+-3·+were unambiguously confirmed by X-ray crystallographic analysis(Fig.4).Their single crystals revealed the distinct propeller-shaped configuration of TPA core,wherein thepara-position on anisyl rings were properly protected by the bulkytert–butyl group.Such configuration and the kinetic protection reasonably elucidated the increased stability of TPA radical cation consisting of more 3,5-di–tert–butyl–4-anisyl groups.Interestingly,the N–C bonds(red numbers in Fig.4)between the N atom and anisyl group in radical cation species became consistently shorter than in the neutral species.Besides,such N–C bond lengths were also much shorter than those between the N atom andpara-bromophenyl group(black numbers in Fig.4).In addition,the C–O bond lengths(blue numbers in Fig.4)were also becoming shorter than in the neutral species.Therefore,the bond length analysis results might provide insight into the electronic structures of thepara–methoxy substituted TPA radical cation,i.e.,its electronic structure was more like a resonance hybrid wherein spin densities could be efficiently delocalized on the anisyl rings(Scheme 1).These findings were also consistent with the aforementioned spin density distributions of 1·+-3·+and the reported theoretical results[46].
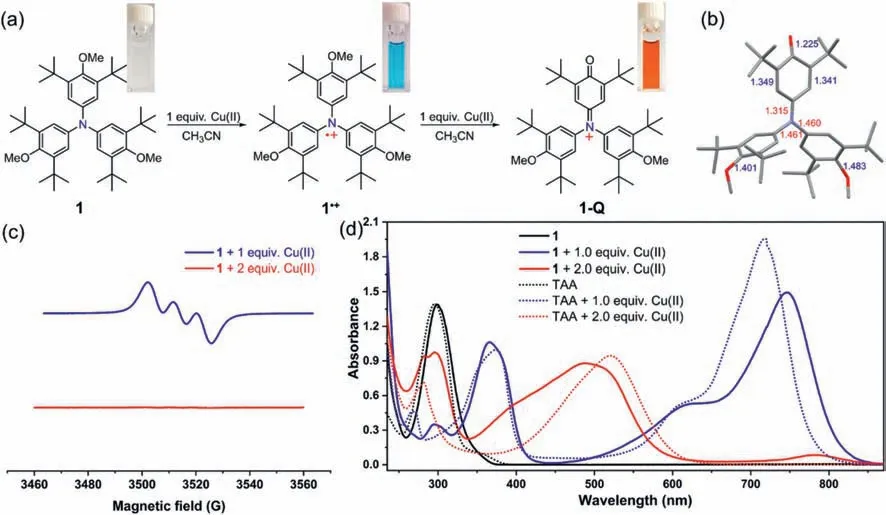
Fig.5.(a)Reaction of 1 with one and two equivalent Cu(ClO4)2·6H2O in acetonitrile.Insert are the photos of the solutions of 1(colorless),1·+(blue)and 1-Q(red).(b)X-ray crystallographic structure of 1-Q(ClO4- anion and hydrogen were omitted for clarity).(c)EPR spectra of 1 after adding 1 equiv.(blue line)and 2 equiv.(red line)Cu(ClO4)2·6H2O in acetonitrile.(d)UV–vis-NIR spectra of 1 and TAA,and their oxidation species in the presence of Cu(ClO4)2·6H2O.
According the results of CV,triphenylamine 1 exhibited two oxidation waves,so it could theoretically be further oxidized to dication species,which was also surveyed in the literature[41].Thus,the stronger oxidizing agent Cu(ClO4)2·6H2O was employed to investigate the second oxidation process since 1 could only be oxidized to radical cation even in the presence of excess AgSbF6.1 could be efficiently oxidized to 1·+by precisely adding one equivalent Cu(ClO4)2·6H2O in its acetonitrile solution,accompanying the formation of a blue solution(Fig.5a)which featured the same UV–vis-NIR and EPR profiles as those of pure 1·+(Figs.5c and d).When adding two equivalent Cu(ClO4)2·6H2O to 1,a new species in red color was generated(Fig.5a),which was EPR silent(Fig.5c,red line)in acetonitrile.Notably,the redox behavior of 1 was virtually the same as that of its analogue tris(4-anisyl)amine(TAA)based on the previous report[47]and our control experiment(Fig.5d and Fig.S4 in Supporting information),appearing to justify the formation of dication species.However,X-ray crystallographic analysis revealed a quinone like structure of 1-Q accompanying demethylation of 1(Fig.5b).Thus,the second oxidation process of 1 was likely to involve an unexpected oxidative demethylation reaction[50,51]rather than dication formation as claimed by Gopidaset al.[47],which was also consistent with its irreversible redox behavior of the second oxidation wave.In order to demonstrate that the changed UV–vis-NIR spectra stemmed from 1-Q,we performed TD-DFT calculation on the 1-Q at the wB97XD/def2-TZVP level of theory,and the results implied that the UV–vis-NIR absorption was really derived from 1-Q(Table S4 in Supporting information).What is more,triphenylamines 2 and 3 can be oxidized to form demethylation product by adding two equivalent Cu(ClO4)2·6H2O as well,whose spectra profiles resembled those of 1(Fig.S5 in Supporting information).The findings may suggest that 4-anisyl substituted TPA and related materials might also undergo such decomposition/degradation when applied at high voltages.
In conclusion,we have synthesized a series of TPA molecules consisting of one,two,or three 3,5-di–tert–butyl–4-anisyl groups and investigated their redox behaviors and corresponding radical cation species.The introduction of anisyl groups significantly reduces the oxidation potential of TPA molecules and enhances the stability of the TPA radical cations.As a consequence,all TPA molecules exhibited low oxidation potentials and could be oxidized to radical cations,wherein 1·+was found to be most stable among them because of the properly steric protection.X-ray crystallographic analysis together with the DFT calculation results disclosed that the electronic structure of 4-anisyl substituted TPA radical cation species was more like a resonance hybrid wherein spin densities could be efficiently delocalized on the anisyl rings,which goes some way to explain why these species are relatively stable.Moreover,an unexpected oxidative demethylation was observed when 1 was further oxidized by a stronger oxidant.These findings are valuable for the understanding of the redox behaviors of 4-anisyl substituted triphenylamines.We hope that our study will shed some light on the design of novel organic functional materials based on triphenylamine derivatives and their radical cation species.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China(Nos.22071061 and 52003081),Shanghai Sailing Pro-gram(No.19YF1412900)and Microscale Magnetic Resonance Platform of ECNU.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.11.054.
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis