
Carbon–sulfur bond formation via photochemical strategies:An efficient method for the synthesis of sulfur-containing compounds
2022-06-20 06:19DoshnYngQiuliYnEnjieZhuJinLvWeiMinHe
Chinese Chemical Letters 2022年4期
Doshn Yng,Qiuli Yn,Enjie Zhu,Jin Lv,Wei-Min He
a Key Laboratory of Optic-electric Sensing and Analytical Chemistry for Life Science,MOE,State Key Laboratory Base of Eco-Chemical Engineering,College of Chemistry and Molecular Engineering,Qingdao University of Science and Technology,Qingdao 266042,China
b School of Chemistry and Chemical Engineering,University of South China,Hengyang 421001,China
ABSTRACT The development of green and convenient methods for C–S bond formation has received significant attention because C–S bond widely occurs in many important pharmaceutical and biological compounds.Recently,visible-light photoredox catalysis has been established as an efficient and general tool for the construction of C–C and C-heteroatom bonds.In this review,we have focused on the research on recent advances in C–S bond formation via visible-light photoredox catalysis,and the growing opportunities they present to the construction of complex chemical scaffolds for applications encompassing bioactive molecules synthesis,synthetic methodology development,and sulfur-containing drugs.We hope that this review will provide chemists with a synthetic tool that will open the door to further development of organsulfur chemistry
Keywords:Visible light C-S bond formation Radical Cyclization Sulfur-containing compouds
1.Introduction
Sulfur is one of the most important nonmetal elements widely occur in all living organisms and nature.Numerous natural products,pesticides,and organic functional materials usually have sulfur-containing frameworks possessing various excellent biological activities and optical properties[1–6].For example,in many biological macro-molecules such as enzymes and transfer ribonucleic acids(tRNAs),sulfur-containing compounds are indispensable which strongly affect their biological activities[7].In addition,organosulfur compounds is the core unit of many commercially available drugs,where they are used for the treatment of many diseases including cancer,diabetes,Alzheimer’s disease,and AIDS(Fig.1)[8].Consequently,the C–S bond forming reaction is of great significance and is a fundamental area of research in organic chemistry.
The classical methods for the construction of C–S bond traditionally involve the following approaches:(a)Nucleophilic substitution reaction between alkyl halides and thiols[9];(b)transitionmetal-catalyzed cross-coupling reactions of thiols with aryl halides,pseudo-halides or arylboronic acids[10–14];(c)transition-metalcatalyzed direct sulfenylation of inert C–H bonds using thiols,sulfenyl halides,disulfides,1-(substituted phenylthio)pyrrolidine-2,5–dione arylsulfonyl cyanides or sulfonyl hydrazides as the thiol source[15–20].Despite great advantages have been made,there are still some limitations,including harsh reaction conditions,strong oxidants,not easily available precursors,and toxic metal salt catalysts.Thus,there is still plenty of room to develop more environmentally benign,simple,and sustainable strategies for the construction of C–S bond.
With the growing demand for environmental protection and the warning of the energy crisis,the development of sustainable and green synthetic strategies for organic transformations is highly desirable.In recent years,visible light-promoted photoredox organic transformations has attracted continuous interest in organic chemistry since the seminal studies from the groups of MacMillan,Stephenson,and Yoon and other groups[21–39].Compared with the traditional methods,visible-light photoredox catalysis,using visible light as a renewable energy source,has been demonstrated as a mild and powerful tool to facilitate activation of organic molecules through a single-electron transfer(SET)process[40–51].This review will summarize the recent advances in the C–S bond forming reactionsviavisible-light promoted reactions.In addition,this review will help readers understand the uniqueness and novelty of photocatalytic technology in sulfenylation reactions(Scheme 1).
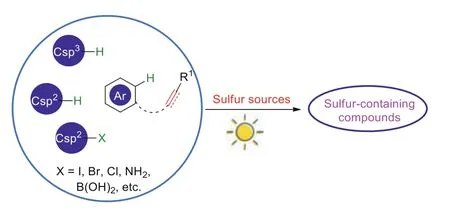
Scheme 1.Overview on the photocatalytic sulfenylation reactions.
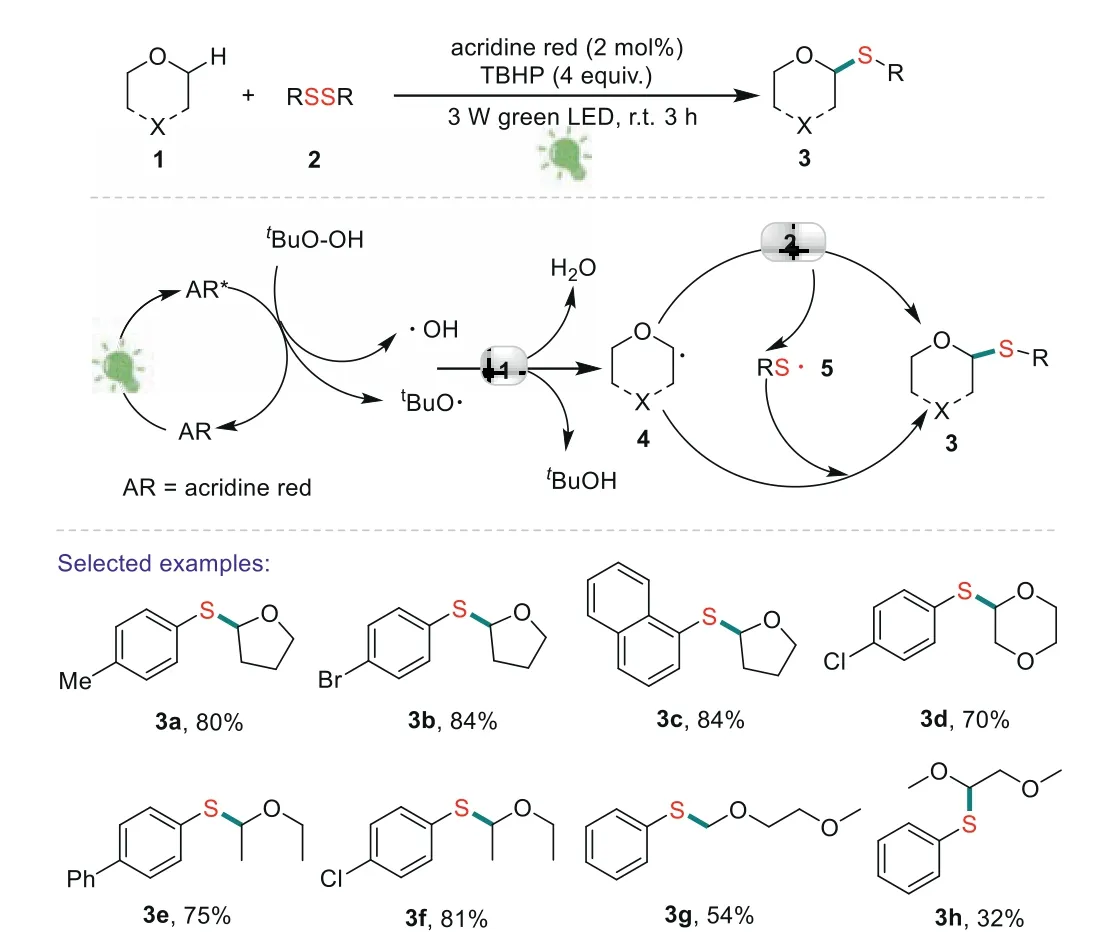
Scheme 2.Direct thiolation at α-C(sp3)-H of ethers with disulfides.
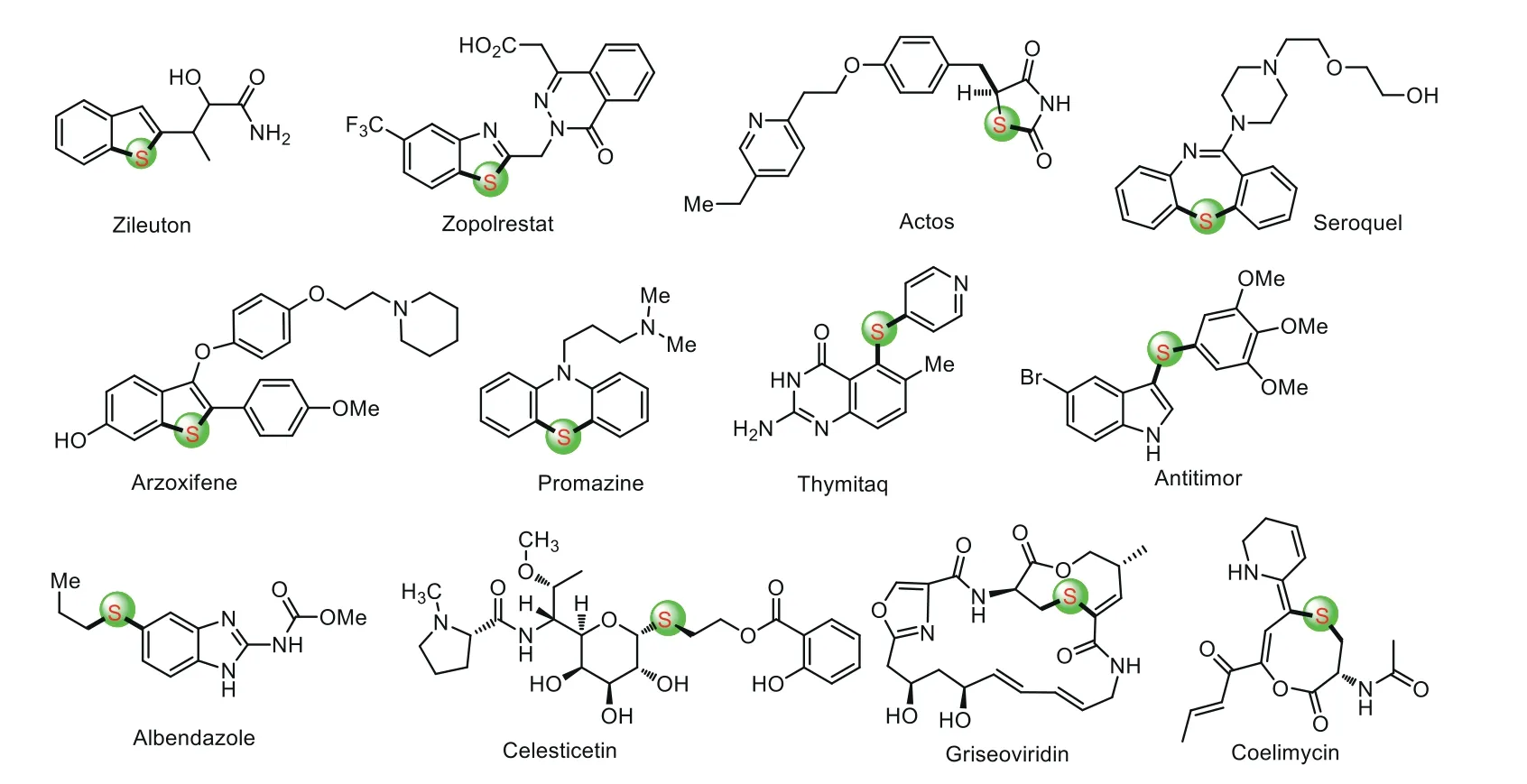
Fig.1.Examples of sulfur-containing compounds in pharmaceuticals.
2.Photochemical C(sp3)-S bond formation
The development of green and efficient methods for the construction of C(sp3)–S bond has gained much attention from the synthetic organic chemists due to their wide occurrence in a large number of natural products and in biologically active molecules.In recent years,C–H activation/functionalization has been established as a powerful technique to construct diverse organic molecules[52–58].Clearly,the direct sulfenylation of C–H bond is more practical and economical.However,the synthesis of C-S bonds through C(sp3)-H functionalization are rather limited.Herein,we summarize the visible-light-mediated C(sp3)-H sulfenylation reactions for the construction of diverse C(sp3)-S bond.
In 2016,Zhu’s group disclosed an elegant and efficient approach toα-arylthioethers through visible light-induced direct thiolationα-C(sp3)-H of ethers with diaryl disulfides(Scheme 2)[59].This is the first example that used acridine red as energy transfer photocatalyst in C-S bond forming reactions.The reactions have the advantages of mild reaction conditions,energy ecology and good functional group tolerance.On the basis of experimental research,they proposed a mechanism that was shown in Scheme 2.Acridine red(AR)was firstly transferred to excited state(AR)*with the irradiation of green LED.Then,the(AR)*interacted witht-BuOOH(TBHP)to generate two crucial radical species,atert–butoxy radical(t-BuO·)and a hydroxyl radical(HO·)viaan energy transfer pathway.Subsequently,tBuO·or HO·abstracted hydrogen fromα-C(sp3)-H of 1,leading to a alkoxyalkyl radical 4.The intermediate 4 reacted with 2 to give the desired product 3 and a new radical 5.Meanwhile,the radical 5 would be trapped by another alkoxyalkyl radical 4 to generate the desired product 3.
Allyl thioethers have been widely used as powerful synthons for the synthesis of thioesters,sulfoxides,sulfones and other sulfur functional compounds.In additions,they also play an important role in the field of biological and pharmaceutical fields[60,61].In 2020,Kim and co-workers initially reported an efficient protocol of the direct allylic C(sp3)-H thiolation induced by visible light photoredox catalysis[62].In the developed method,visible light photoredox catalysis was used as an efficient promoter to induce selective HAT(a hydrogen atom transfer)at the allylic position.A broad range of diaryl disulfides and olefins were tested,and provided the desired products in good to excellent yields.Based on the experimental results and in accordance with theoretical studies,a plausible pathway for this thiolation reaction was proposed in Scheme 3.Firstly,thiyl radical 9 could be generated through S-S homolytically cleaved by irradiation of the blue LED.The formed thiyl radical 9 could participate in either addition to 7 to give a thiol-ene radical adduct 11 or HAT from the allylic position to form an ally radical 10.Subsequently,the resulting thiophenol was deprotonated by hydroxide,which prevented further hydrogen transfer to 11,leading to the irreversible HAT process.Then,the intermediate 10 underwent single-electron oxidation by Ir(III)*and gave allyl cation 13.Finally,allyl cation 13 reacted with the 12 produced the desired product 8.
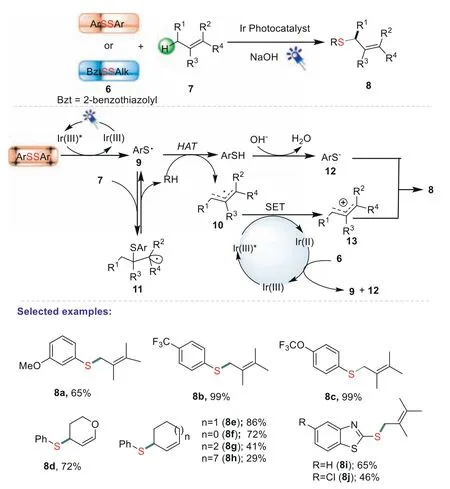
Scheme 3.Direct allylic C(sp3)-H thiolation with disulfides.
In 2019,Jiet al.revealed a photocatalytic aerobic coupling of mercaptobenzimidazoles with carbonyls[63].Advantages of this photocatalytic reaction include using O2as the green oxidant,and easily available rose bengal as the photocatalyst.Based on the control experiments,the following mechanism was proposed.Initially,(RB)was converted into the excited RB*under visible-light irradiation conditions.Then,thiol 14 reacted with RB*through a SET to afford the thiyl radical ion 17,which underwent a deprotonation to form radical 18.The addition of 18 to enol 19 leads to alkyl radical 20.Finally,a SET process and deprotonation pathway took place again to deliver sulfide intermediate 21,which subsequently converted to the products 16 through nucleophilic annulations and workup(Scheme 4).
Furthermore,visible light induced decarboxylation coupling reactions can also be used as an efficient approach to build C-S bonds.In 2018,Xiao and co-workers reported a novel and convenient visible-light-promoted decarboxylative coupling reaction betweenN-hydroxyphthalimide esters and diaryl disulfides(Scheme 5)[64].This developed protocol has some merits including the use of broad substrate scope,mild conditions,gram-scale synthesis,and high product yields,which demonstrates the high efficiency and practicality.A plausible catalytic cycle was proposed.Firstly,the reaction was induced by the SET between DIPEA and Ru*(II).Next,redoxactive ester 23 was reduced by the Ru(I)species to give alkyl radicals 27 with releasing CO2.Subsequently,the active alkyl radical 27 reacted with diaryl disulfide 24 to generate the desired product 25.
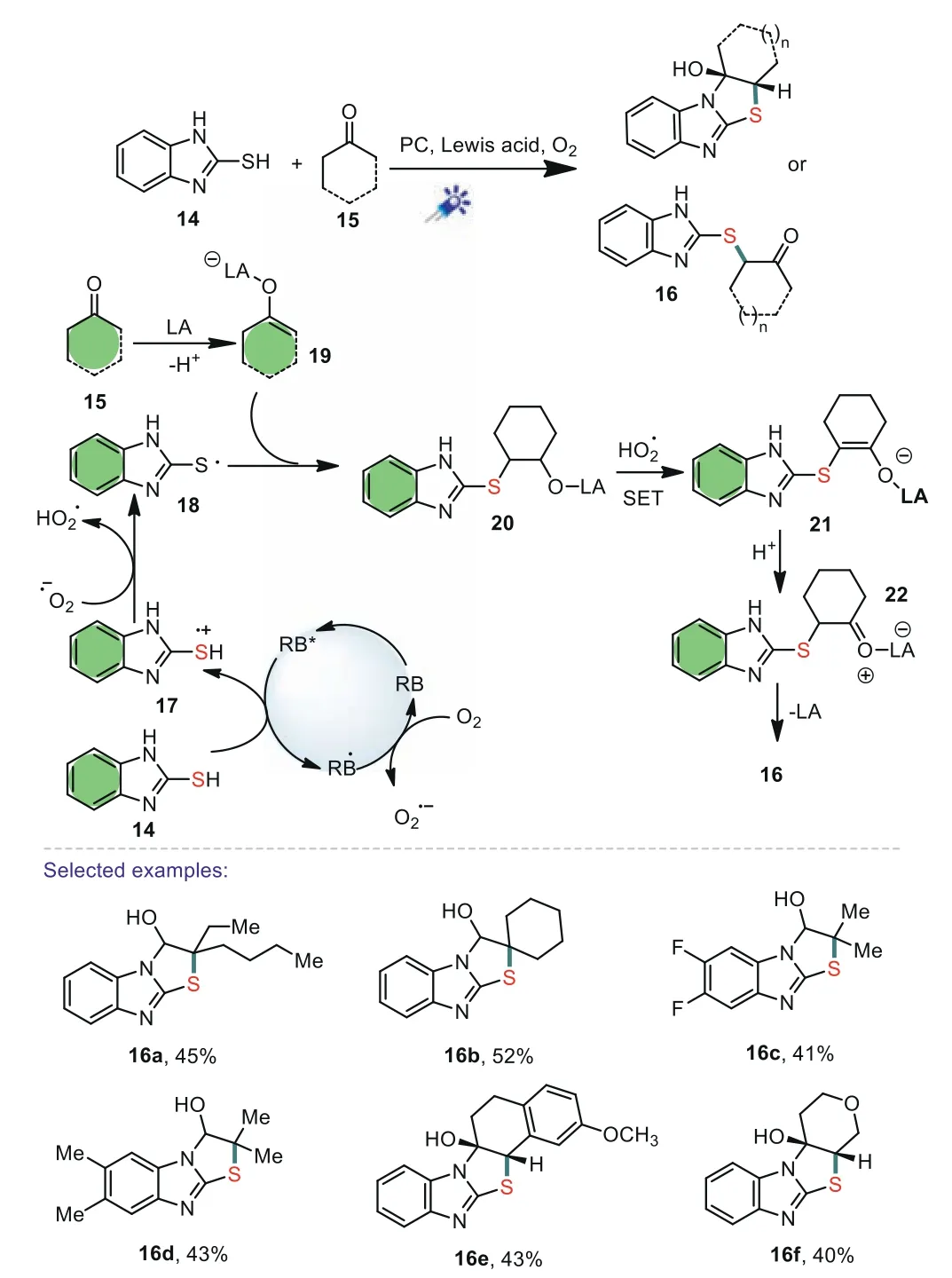
Scheme 4.Proposed mechanism for the direct transformation.
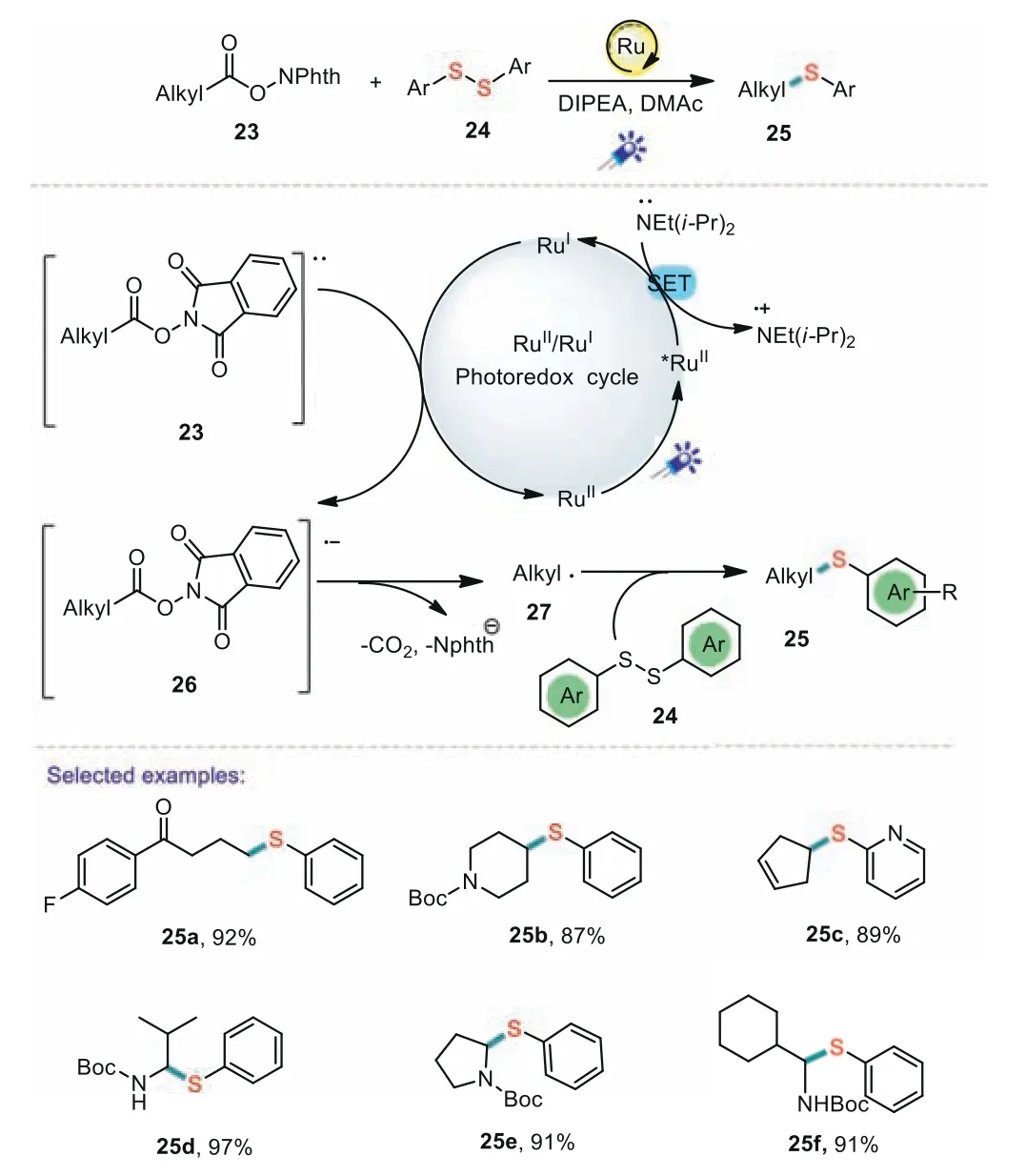
Scheme 5.Decarboxylative coupling of redox-active esters with disulfides.
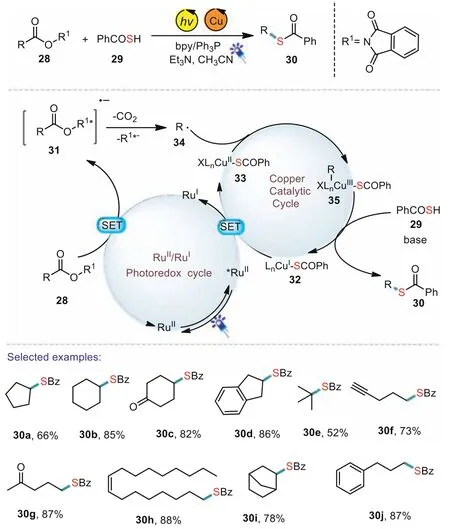
Scheme 6.Photoinduced decarboxylative thiolation tothioesters.
In 2020,Liao’s group also demonstrated an efficient decarboxylative thioesterification reaction of redoxactive esters by visible light and copper catalysis(Scheme 6)[65].A various of carboxylic acids,including primary,secondary,and tertiary carboxylic acids,as well as several pharmaceuticals and natural products were well compatible in the present transformation.In addition,the carboxylic groups of drugs such as Indometacin,Gemfibrozil,and Fenbufen could be easily converted to the corresponding thioesters.Based on several control experiments,a possible reaction mechanism was proposed.Under light irradiation,the photocatalyst[RuII]was firstly excited and then accepted an electron from LnCu(I)species 32 to afford the corresponding highly reducing[RuI]and the key Cu(II)species 33.Subsequently,a single electron transfer from[RuI]to the redox-active ester 28 gave the alkyl radical 34,which could react with 33 to generate the key intermediate 35.Then 35 reacted with 29 through a reductive elimination pathway to deliver the desired thioester products 30.
In 2019,Wei and co-workers reported a thiolation reaction from carboxylic acids through the combination of photoredox catalysis and nickel catalysis(Scheme 7)[66].This is the first example of a decarboxylative thiolation reaction from carboxylic acids using benzenesulfonothioates as odorless thiolation reagents.The proposed mechanism is shown in Scheme 7.First,through the SET process,photoredox catalysis reacted with 36 and gave the alkyl radical 39.Then,the generated radical 39 reacted with 37 to form the product 38.Meanwhile,the reactive Ni(0)reacted with 37 to generate Ni(II)species 41 by oxidative addition to the N–S bond.Subsequently,41 coupled with radical 40 to produce the products 38 through a reductive elimination process.
The thiol-ene coupling(TEC)reaction has attracted significant attention during the past few years for the formation of C(sp3)–S bonds which could be successfully used to construct a wide range of organosulfur compounds,particularly in the fields of materials,polymers,and drugs[67].In 2014,Tyson and co-workers disclosed a new strategy for highly efficient radical thiol-ene conjugation of various alkenes and thiols using the thiol component as the limiting reagent[68].This method exhibits remarkable functional group tolerance across a wide scope of substrates.Aliphatic thiols with free hydroxyl and secondary thiols were well tolerated in the present photocatalytic transformation.Control experiments indicated thatp-toluidine serves as a redox mediator that was capable of catalyzing the otherwise inefficient photooxidation of thiols to the key thiyl radical intermediate.Notably,glutathiones were also compatible in the reaction and gave the corresponding biologically active molecules in good yields(Scheme 8).
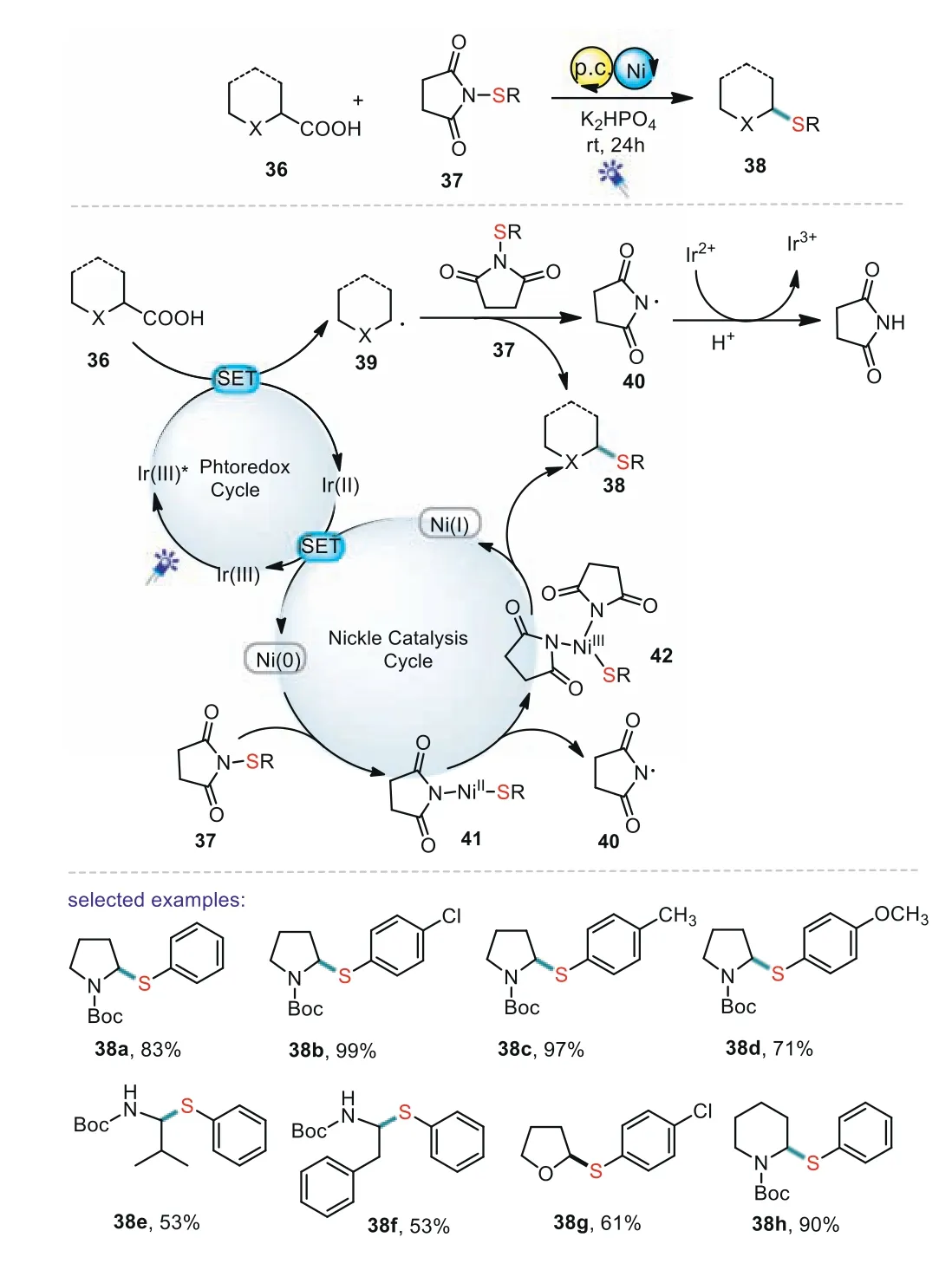
Scheme 7.Decarboxylative sulfenylation of amino acids.
Subsequently,Bhat’s group reported an elegant visible lightpromoted thiol-ene reactions using titanium dioxide as a photoredox catalysis(Scheme 9)[69].The reaction proceeded under mild conditions and tolerated a variety of functional groups(ester,nitrile,halogen,silane,and alcohol)giving the corresponding products in good yields.Based upon the photo-excitation of electrons to the conduction band of the titania catalyst,a possible mechanism for the transformation is proposed.Firstly,the thiol could reductively quench the hole and delivered a thiyl radical cation,which subsequently lost a proton to afford a thiyl radical.Next,the thiolene cycle could be initiated by the thiyl radical through addition to an alkene and formation an alkyl radical,which reacted with thiol to give a another thiyl radical.In the catalytic cycle,oxygen worked as a sacrificial electron acceptor,promoting reaction effi-ciency by reducing hole-electron recombination in the titania.
In 2015,Fadeyi and co-workers have also reported an effi-cient visible-light-promoted radical thiol-ene reactions using bismuth oxide as the photoredox catalyst(Scheme 10)[70].The developed method delivered the desired thioether products in good yields,and various functional groups(including esters,alcohols,Boc-protected amines,boronic pinacol esters,carboxylic acids,and pyridine-based heteroaromatics)are well tolerated under the optimized reaction conditions.A similar mechanism was proposed as in Scheme 11.
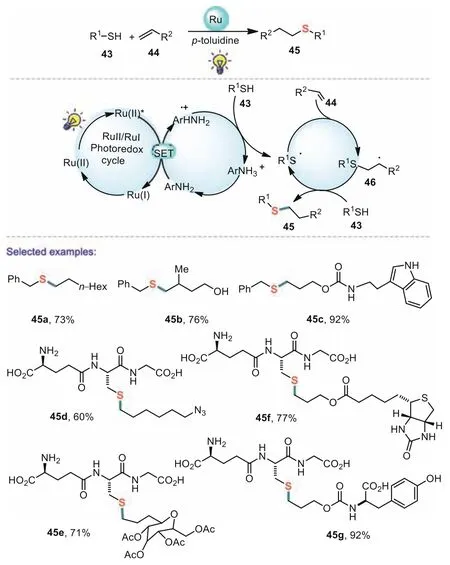
Scheme 8.Redox mediated radical thiol-ene reaction.
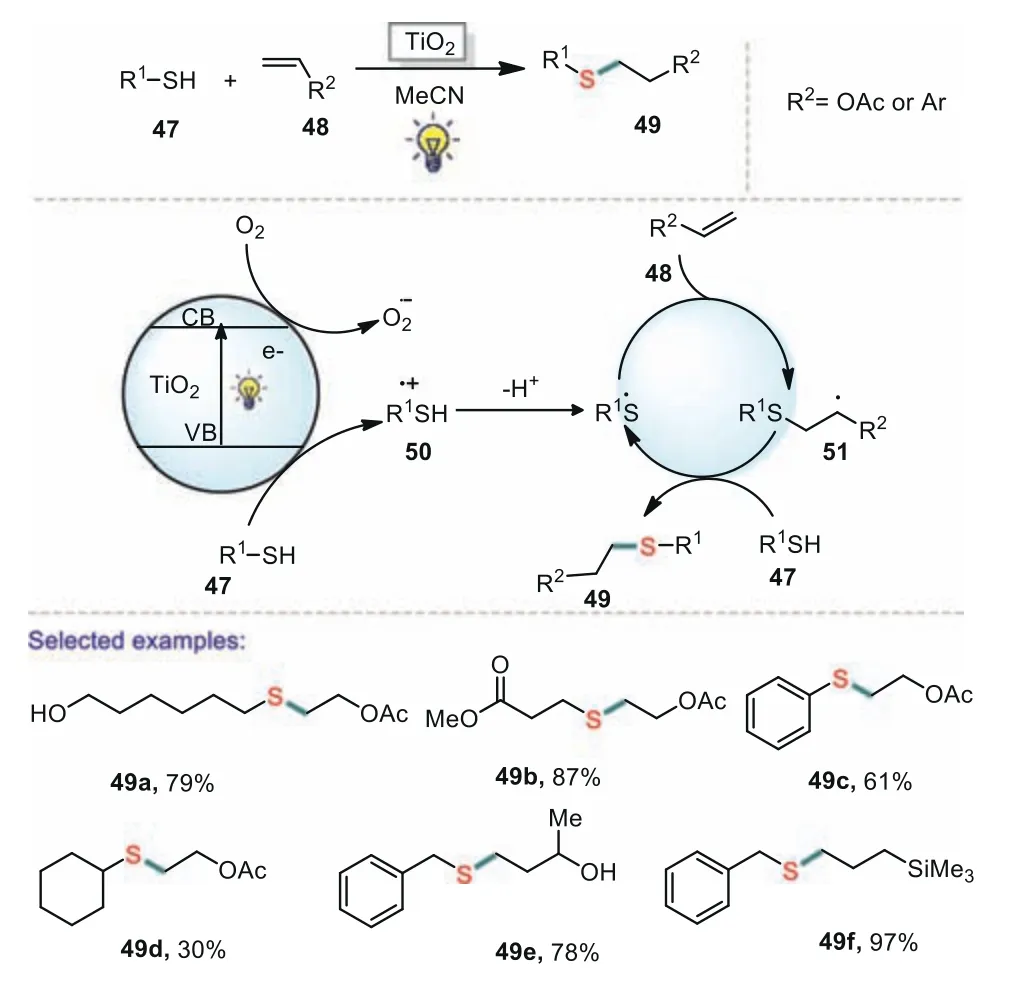
Scheme 9.Visible light promoted thiol-ene reactions using titanium dioxide.
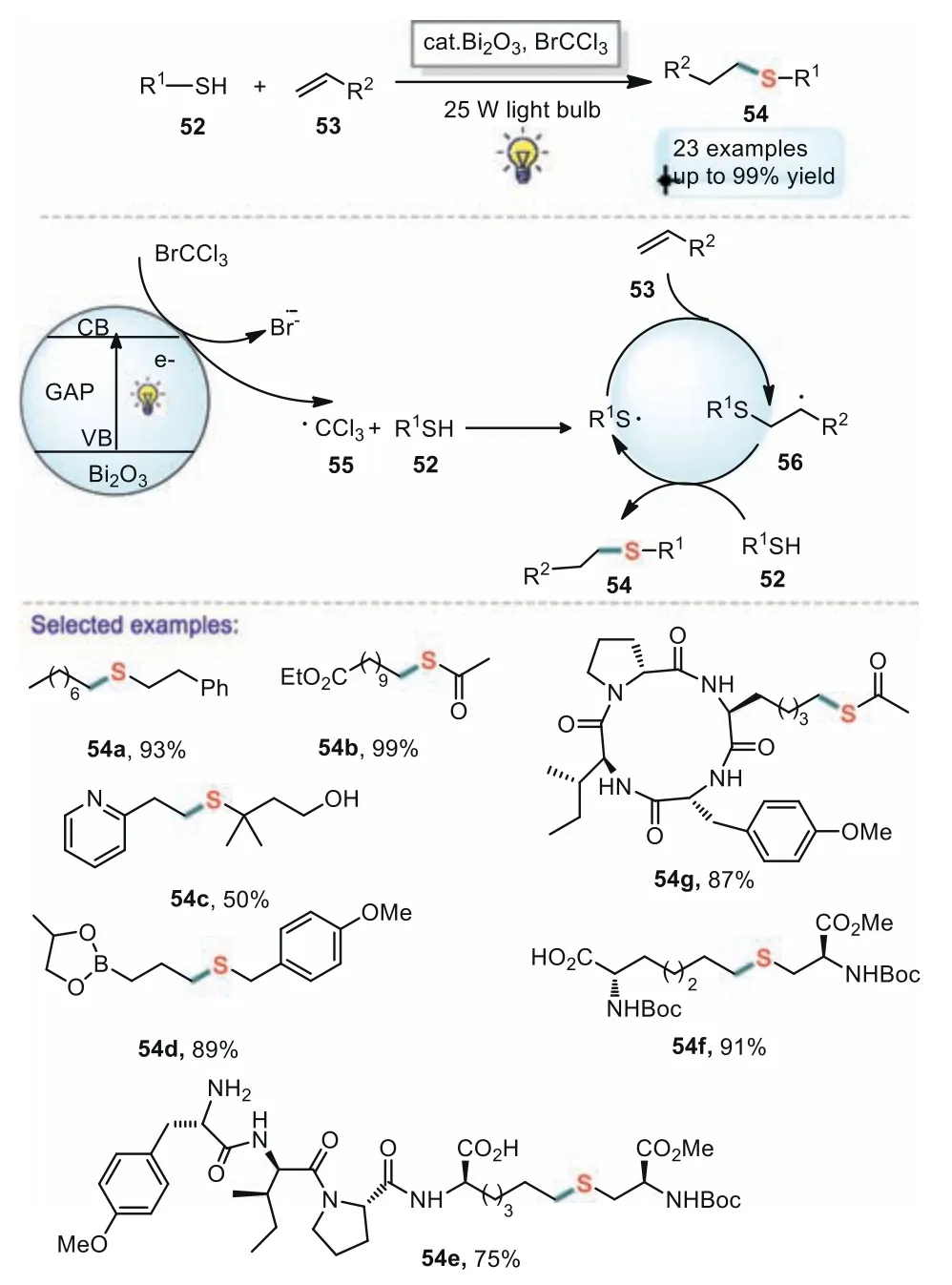
Scheme 10.Visible light promoted thiol-ene reactions using bismuth oxide.

Scheme 11.Visible light promoted thiol-ene reactions using phenylglyoxylic acid.
The combination of photocatalysis and organocatalysis is a unique and useful method in photocatalytic reactions.In 2017,Limnios and co-workers developed a simple and efficient organocatalytic photoinduced strategy for the thiol-ene coupling(TEC)reaction between a variety of thiols and olefins using phenylglyoxylic acid as the catalyst-initiator(Scheme 11)[71].Control experiments indicated that a chain propagation mechanism was dominant.This developed approach might find wide applications in the field of glycoside and peptide modifications.
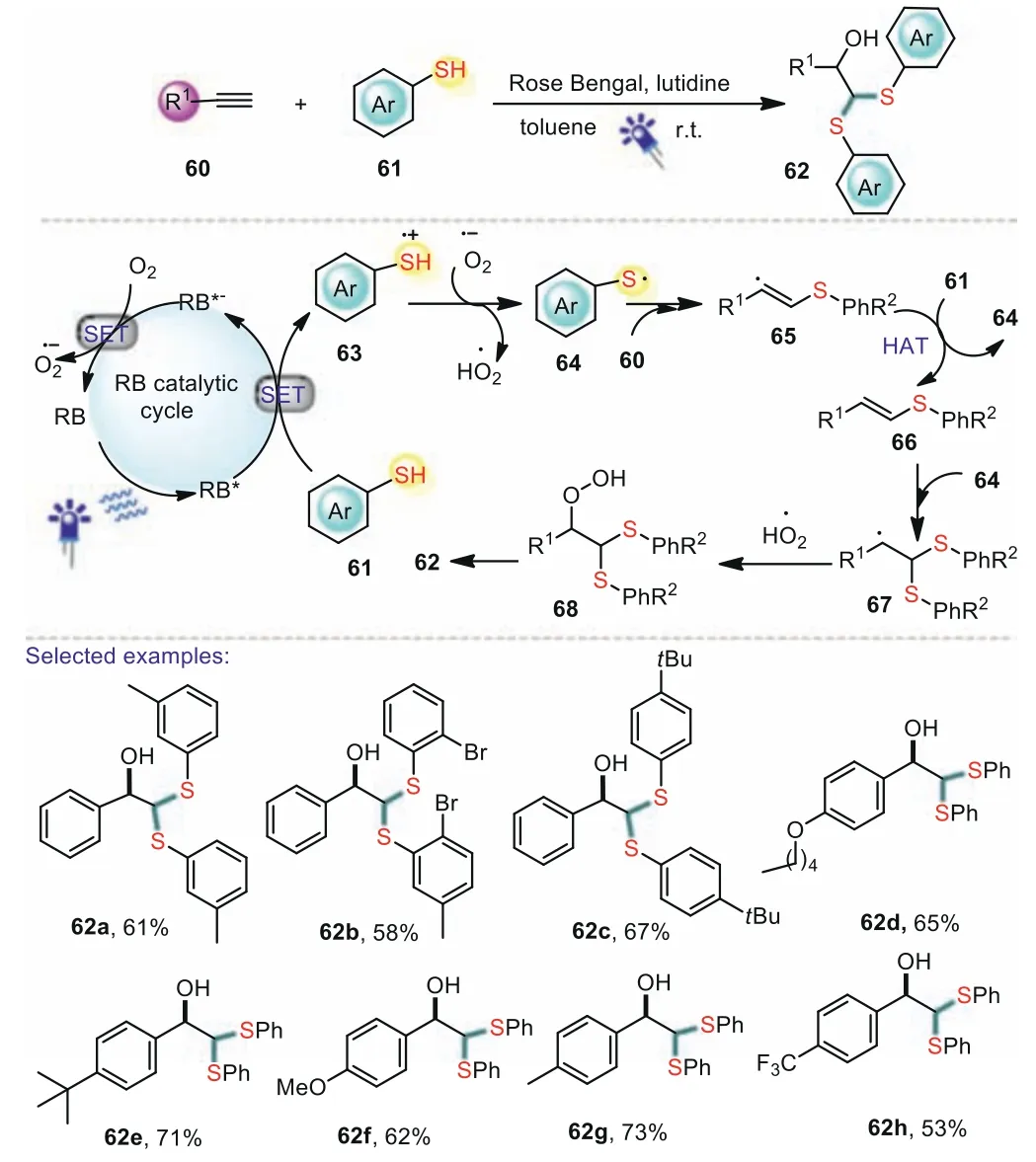
Scheme 12.Photoredox-mediated synthesis of β-hydroxydithioacetals from terminal alkynes.
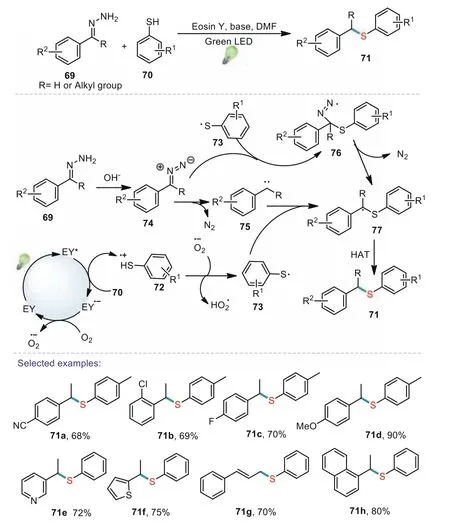
Scheme 13.Visible-light induced sulfenylation of hydrazones leading to thioethers.
Besides the thiol-ene reactions,the alkynes are usually used as radical acceptors to react with S-radicals.In 2020,Shah and coworkers developed a single-step photocatalytic thiolation protocol for the synthesis ofβ-hydroxydithioacetals from terminal alkynes[72].The reaction was achieved by air oxidation under blue LEDs irradiation with high reaction efficiency,and could be extended to aliphatic and aromatic alkynes.The electronic effect of substituents in aromatic alkynes had little effect on the reaction.In addition,thioacetals bearing hydroxyl groups could also be applied for the construction ofα–hydroxy aldehydes which serve as building blocks in the synthesis of various natural products.A plausible mechanism was shown in Scheme 12.Firstly,the photoexcited*RB was reductive quenched by thiophenol 61viaa SET process to generate the thiyl radical cation 63,that subsequently deprotonated to form thiyl radical 64.Then,the thiyl radical 64 was added to alkyne 60 to give the reactive vinyl radical 65,which underwent hydrogen atom transfer with 61 providing vinyl sulfide 66.Subsequently,vinyl sulfide 66 reacted with 64 to afford disulfide radical species 67.Finally,the hydroperoxide radical reacted with 67 and further cleavage of hydroperoxide gives 62.
Hydrazones used as precursors to the diazo compounds and carbene sources,have been regarded as important synthons for various organic transformations[73].A diverse array of C–C and C–heteroatom bonds with good regio-and stereoselectivity could be constructed under mild conditions using hydazones as building blocks.In 2019,an efficient and simple visible-light-promoted Eosin Y catalyzed sulfenylation of hydrazones has been reported by Chand’s group[74].This method has a wide range of substrate scope and various functional groups were well tolerated in the oxidative transformation.Control experiments indicated that both the light irradiation and photocatalyst were essential for the sufenylation pathway.For the reaction mechanism,Eosin Y was firstly converted into the excited state under green-light irradiation.The photo-excited Eosin Y was presumed to oxidize 70 to the corresponding thiyl radical cation 72.Then,the reduced Eosin Y radical anion reacted with O2to generate the superoxide radical anion(O2-).Subsequently,the superoxide radical anion deprotonates 72 to form the thiyl radical 73 and hydroperoxyl radical.Hydrazone acts as a precursor to the diazo compound,which on addition with the 73 gives the intermediate 76.Then the intermediate 76 underwent dediazotization to generate the C-centered radical 77.Alternatively,hydrazone could convert into intermediate carbene 75 in the presence of hydroxide ion.The intermediate carbene 75 subsequently coupled with the 73 to generate the C-centered radical 77,followed by hydrogen atom transfer(HAT)to provide the product 71(Scheme 13).
4-Alkyl-1,4-dihydropyridines(DHPs)can be readily prepared from aldehydes in one step with high functionalization levels.Since the pioneering work of Nishibayashi,C–C bond cleavage of DHPs has been used as an robust tool to construct C-C and C-heteroatom bonds[75].In 2020,Ji and Wang’s group developed a visible-light-induced cross-coupling of 4-alkyl-1,4-dihydropyridines(DHPs)with thio-/selenium sulfonates for the synthesis of sulfides,sulfoxides,and selenides[76].Importantly,the developed method was extended to the construction of a class of biologically selenylated or thiolated glycosides.In addition,sulfoxides were also obtained chemoselectivelyviaa variation of the atmosphere under photocatalyzed reaction conditions.As for proposed mechanism is shown in Scheme 14.The photocatalyst 4CzIPN(PC)prompted the SET of the DHPs to produce an alkyl radical 84 and pyridine 85.Next,the alkyl radical 84 reacted with 80 to generate a sulfone radical 86.Through reductive SET process,86 furnished benzosulfinic acid anion 87 which subsequently afforded the benzosulfinic acid through protonation pathway.On the other side,1O2could be produced from an energy-transfer pathway between PC*and3O2,which could oxidize the sulfide to the corresponding sulfoxide 82.
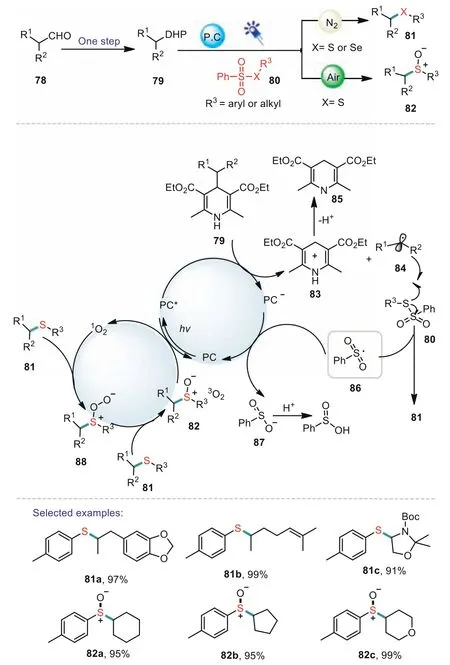
Scheme 14.Visible-light-promoted cross coupling of 4-alkyl-1,4-dihydropyridines with thio-/selenium sulfonates.
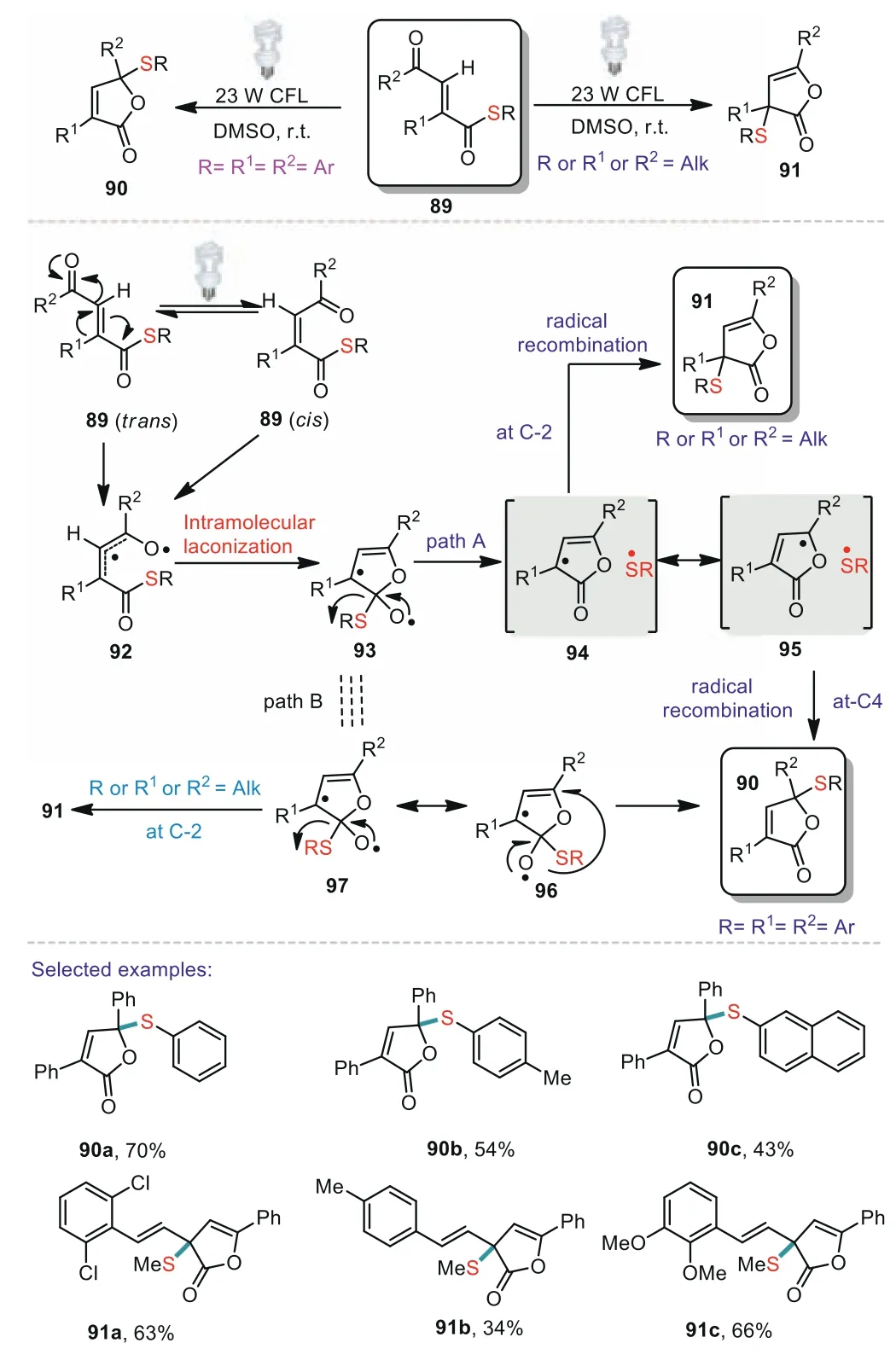
Scheme 15.Visible light induced chemoselective thiyl radicals migration.
Thiyl radical migration plays an important role in the construction of C(sp3)-S bond.In 2016,Naskar reported an unprecedented visible light promoted chemoselective rearrangement ofγ-keto acrylate thioestersviaelusive thiyl radical migration(Scheme 15)[77].Various ofγ-keto acrylate thioesters bearing arylvinyl groups atα-position could afford corresponding products in good yields.In addition,thioesters based on aromatic or alkyl thiols were well tolerated in the present transformation.Two possible pathways were proposed for the thiyl radical migration transformation.Pathway A involved a radical cage process,whereas pathway B proceededviaa direct thiyl radical migration.By visible light irradiation,trans-thioester 89 suffered isomerization leading tocisthioester 89viastabilized radical allylic intermediate 92.Then 92 underwent intramolecular radical lactonization to afford intermediate 93.In the path A,intermediate 93 could afford radical pairs 94 or 95 within a solvent cageviaallylic migration which then suffered radical recombination either at C-2 or at C-4 depending upon the substrates to yield the 90 or 91,respectively.Alternatively,the radical reaction might also occurviaa direct thiyl radical migration from intermediate 93(path B).The intermediate 93 could undergo either 1,2-thiyl radical migration to afford the 91 or 1,4-thiyl radical migration when the substrate was fully substituted by aromatics to generate 90(Scheme 15).
3.Photochemical C(sp2)-S bond formation
3.1.Aryl/heteroaryl C(sp2)-H sulfenylation
The development of catalytic methods to generate C(sp2)–S bonds remains an important endeavor within the chemical sciences,due to the ubiquity of the aryl thioether motif in pharmaceuticals and organic materials.Synthesis of aryl sulfidesviaC-H functionalization under metal-free[78–82],or transitionalmetal-catalyzed conditions with diverse sulfur sources such as arylsulfonyl chlorides,sodium arylsulfinates,thiolphenol,disulfides,sulfinicacids,and arylsulfonyl hydrazides have attracted considerable attention from the synthetic community[83–86].3-Substituted indoles compose an important framwork in agrochemicals,natural products,and functional materials[87].Thus,the development of simple and efficient synthetic approaches to 3-substituted indoles has been of continuous interest in organic chemistry.In 2012,Chenet al.developed an efficient visible lightinduced 3-sulfenylation ofN-methylindoles with arylsulfonyl chlorides under mild conditions[88].The products were obtained in 33%–68% yield with good functional groups tolerance.The position of the substituent on the benzene ring of arylsulfonyl chlorides will affect the efficiency of the reaction.Steric hindrance ofo-methyl made the reaction sluggish,with longer reaction time and lower yield.After the preliminary mechanistic study,a possible mechanism was proposed by the authors.The substrate indoles 98 reacted with Ru(bpy)32+*to generate intermediate 101viareductively quenching pathway.Then three steps of SET process afforded critical intermediate 104.Finally,98 attacked 104 to produce 105,which was transformed into the desired product 100 with releasing a proton(Scheme 16).
In 2017,Fan’s group developed an elegant visible-light induced transformation of indoles into 3-sulfenylindoles using easily available thiophenols as sulfenylating agents[89].Diverse thiophenols and indoles could be efficiently converted to the corresponding 3-sulfenylindoles under mild conditions.The reaction employs readily available and inexpensive rose bengal as a photocatalyst and air as the green oxidant.Control experiments indicated that a radical pathway might involved in the present transformation.A possible catalytic cycle was proposed as shown in Scheme 17.
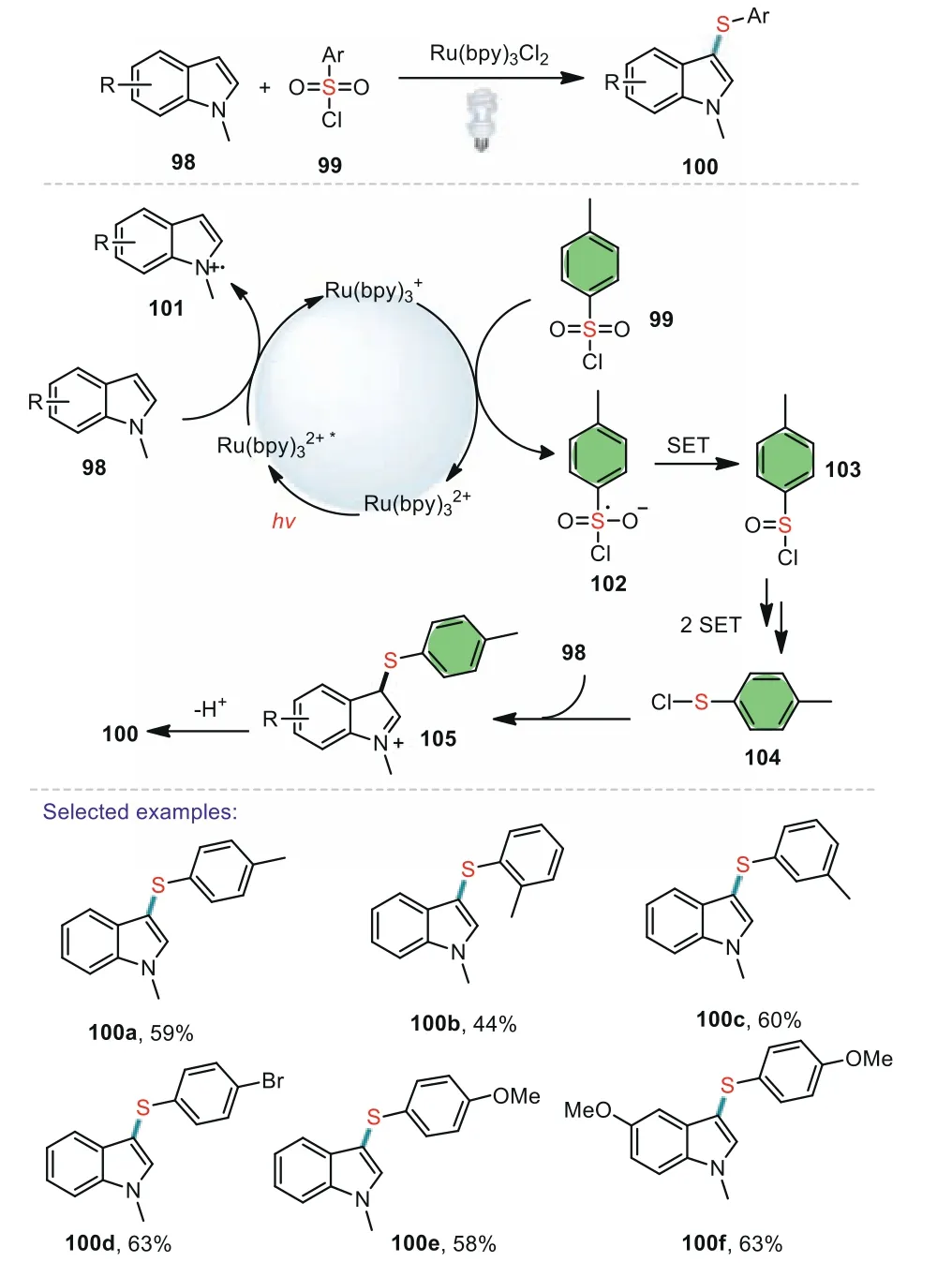
Scheme 16.Visible light-induced 3-sulfenylation of N-methylindoles.
In 2017,Ye and co-workers reported an efficient method for the synthesis of 3-arylthioindoles from indoles and diaryl disulfides under visible light conditions[90].The protocol was efficiently promoted by the catalytic amount of sodium iodide and the desired products were obtained in moderate to good yields with a wide range of substrates scope.Control experiments indicated that the generation of arylthiyl radical through the homolysis of diaryldisulfides was considered as the key initiation step.A possible catalytic cycle was proposed as shown in Scheme 18.The diphenyl disulfide 114 was initially activated by visible light irradiation.A single electron transfer from iodide to the activated diphenyl disulfide provided a diphenyl disulfide anion radical 116.Then 116 generated a thiophenol anion 118 and a phenylthiyl radical 117.The coupling of iodine radical and 117 afforded arylsulfenyl iodine 119.Finally,electrophilic substitution with the indole provided the 3-arylthioindole product 115.
A similar strategy was developed by the group of Kumar.In 2019,they developed a photocatalyst and transition metal-free visible light promoted method for the formation of C–S bond that provided 3-arylthioindoles using oxygen as a benign oxidant(Scheme 19)[91].The developed method is amenable and sustainable for potentially operational procedures,and various diarylsulfides and indoles are well tolerated in the present tranformation.A mechanistic understanding by UV–visible,EPR spectroscopy,and cyclic voltammetry demonstrates that light promotes electron transfer from the electron rich arene to oxygen providing an indole cation 125 and a superoxide radical anion.Then,treatment 125 with diaryl disulfides 122 to give intermediate 127 which subsquently reacts with anion radical of O2·-leading to the desired product 123.
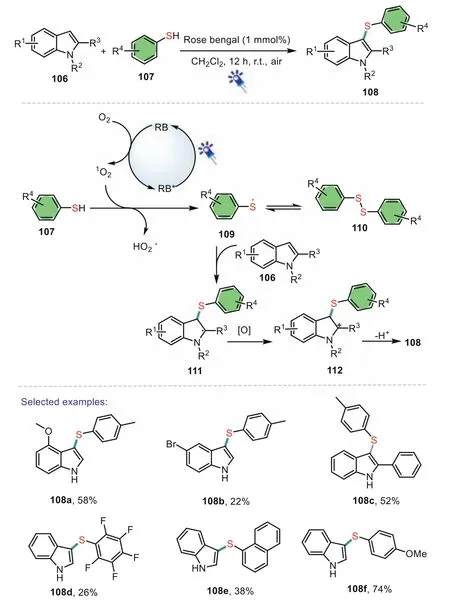
Scheme 17.Visible light-induced C-3 sulfenylation of indoles with thiophenols.
In 2020,Hazarika and Barman.reported a feasible and sustainable protocol for the sulfenylation of electron rich aromatic compounds,active methylene derivatives,and indoles under visible light conditions(Scheme 20)[92].The developed method features a wide range of functional group tolerances.Thiols bearing electron-donating groups and the electron-withdrawing groups reacted with indoles to afford the corresponding products in good yields.Mechanism investigation indicated that the transformation involves a radical pathway.Firstly,CS(Cecosporin)was initially activated by visible light irradiation and reached to its excited state species CS*.Subsequently,the CS*reduced indole 128 to produce radical intermediate 134 and cercosporin radical cation CS·+.The CS·+served as a strong oxidant to oxidize thiophenol to afford the thiol radical cation 135.Finally,the thiol radical cation 135 underwent deprotonation to produce the stabilized phenylthiyl radical 136 which finally reacted with the radical intermediate 134 resulting in the formation of the corresponding product 131.
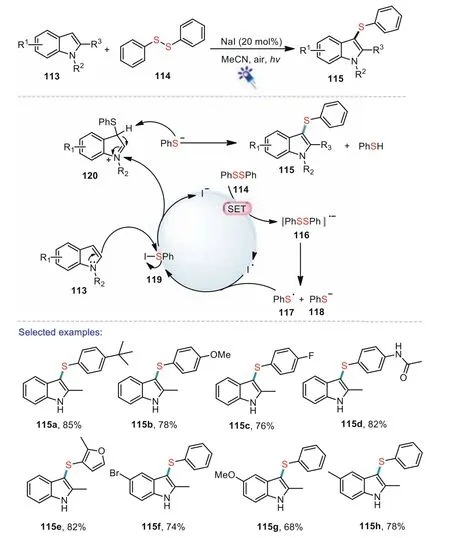
Scheme 18.Visible light-induced C-3 sulfenylation of indoles with diaryl disulfides.
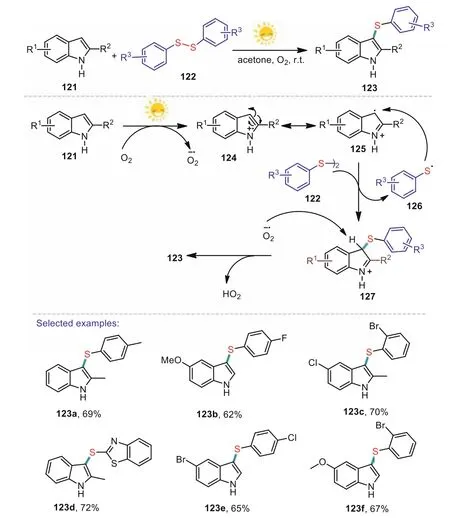
Scheme 19.Photocatalyst-free,visible light-induced C-3 sulfenylation of indoles.
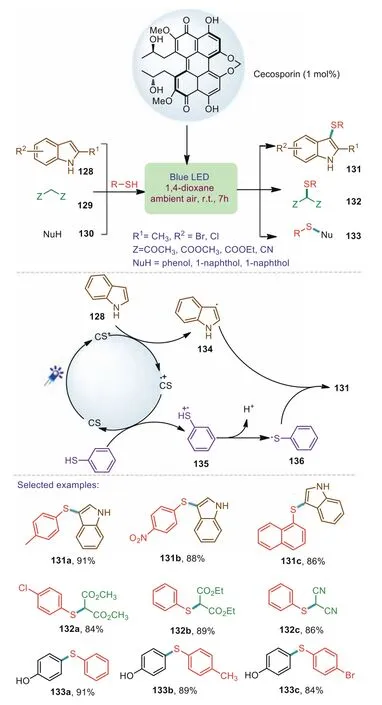
Scheme 20.Visible-light induced sulfenylation of electron-rich compounds with thiols.
Imidazoheterocycles widely occur in numerous biological compounds possessing excellent biological activities[93].In recent years,significant progress has been made in the discovery of efficient methods for the synthesis and functionalization of imidazo[1,2-a]pyridines.In 2017,Sun’s group succeeded in the utilization of arylsulfinic acids as odorless sulfur reagents for the construction of imidazo[1,2-a]pyridine skeletonsviadirect sulfenylation of sp2C–H bonds reactions under visible-light promoted conditions(Scheme 21)[94].In this reaction,diverse sulfinic acids,with either electron-withdrawing or electron-donating groups,were all efficiently converted to their corresponding C-3 sulfenylated imidazoheterocycles in good yields.In addition,imidazopyridines with the electron-donating as well as electron-withdrawing were well tolerated,showing no obvious electronic effect in this transformation.Based on the control experiments,it is reasonable to assume that the photocatalyst Eosin B was excited by visible light irradiation leading to the excited species Eosin B*,which then underwent SET to TBHP,generating thetert–butoxyl radical 140 and a hydroxyl anion 141.The 140 abstracted a hydrogen from 138 to afford the sulfonyl radical 142.Then,the sulfonyl radical 142 reacted with arylsulfinate to provide the corresponding sulfinyl radical 145,which was reduced by H2O ort-BuOH to form the thiyl radical 146.Selective addition of the thiyl radical to the imidazoheterocycle formed the carbon-centered radical 146,which could be further transformed into the carbocation intermediate 147 SET with Eosin B·+.Finally,the sulfonic acid anion attacked theβ-H of intermediate E,resulting in the desired product 139.

Scheme 21.Visible light-induced C–H sulfenylation using sulfinic acids.
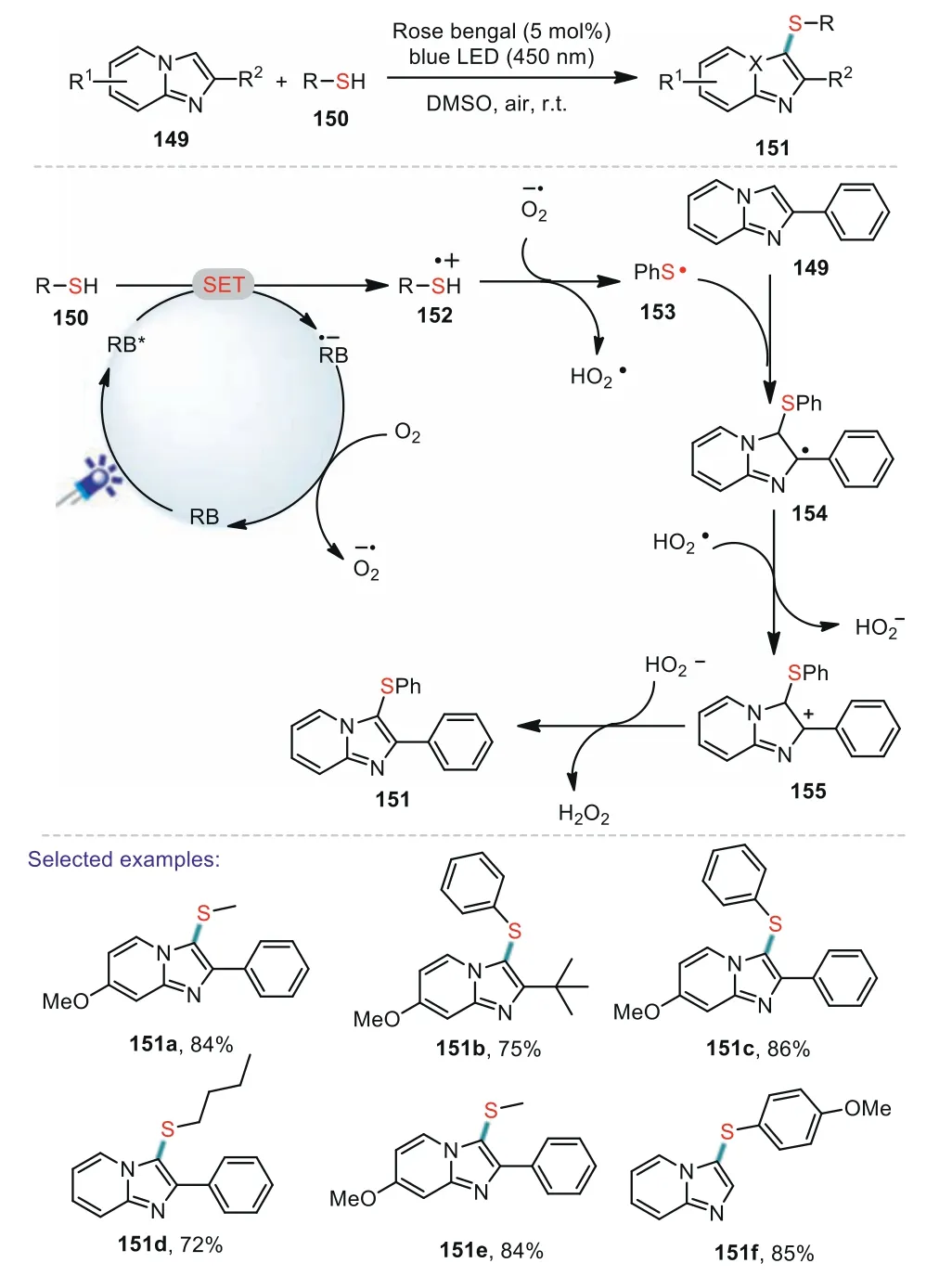
Shortly thereafter,a similar strategy was developed by the group of Rahaman.An efficient and sustainable visible light induced direct C–H bond sulfenylation of various imidazo[1,2-a]pyridines has been achieved to produce the 3-sulfenylimidazopyridines in good yields(Scheme 22)[95].Notably,besides aryl thiols this developed method is also applied to alkyl thiols such as methanethiol and butanethiol.A plausible reaction pathway for the photocatalytic sulfenylation of imidazo[1,2-a]pyridine was proposed in Scheme 22.Firstly,RB*was generated from rose bengal(RB)under the visible-light irradiation of the blue LED lamp.Subsequently,a SET from thiol 150 to RB*afforded the radical cation 152 and RB·-.Then,RB·-was oxidized by air to generate the ground state rose bengal and O2·-.The deprotonation of 152 by O2·-led to the formation of stabilized thiyl radical 153.The generated 153 reacted with 149 to provide the radical intermediate 154.Finally,154 was oxidized to intermediate 155 along with the generation of HOO·-.The deprotonation of 155 generated the desired product 151.
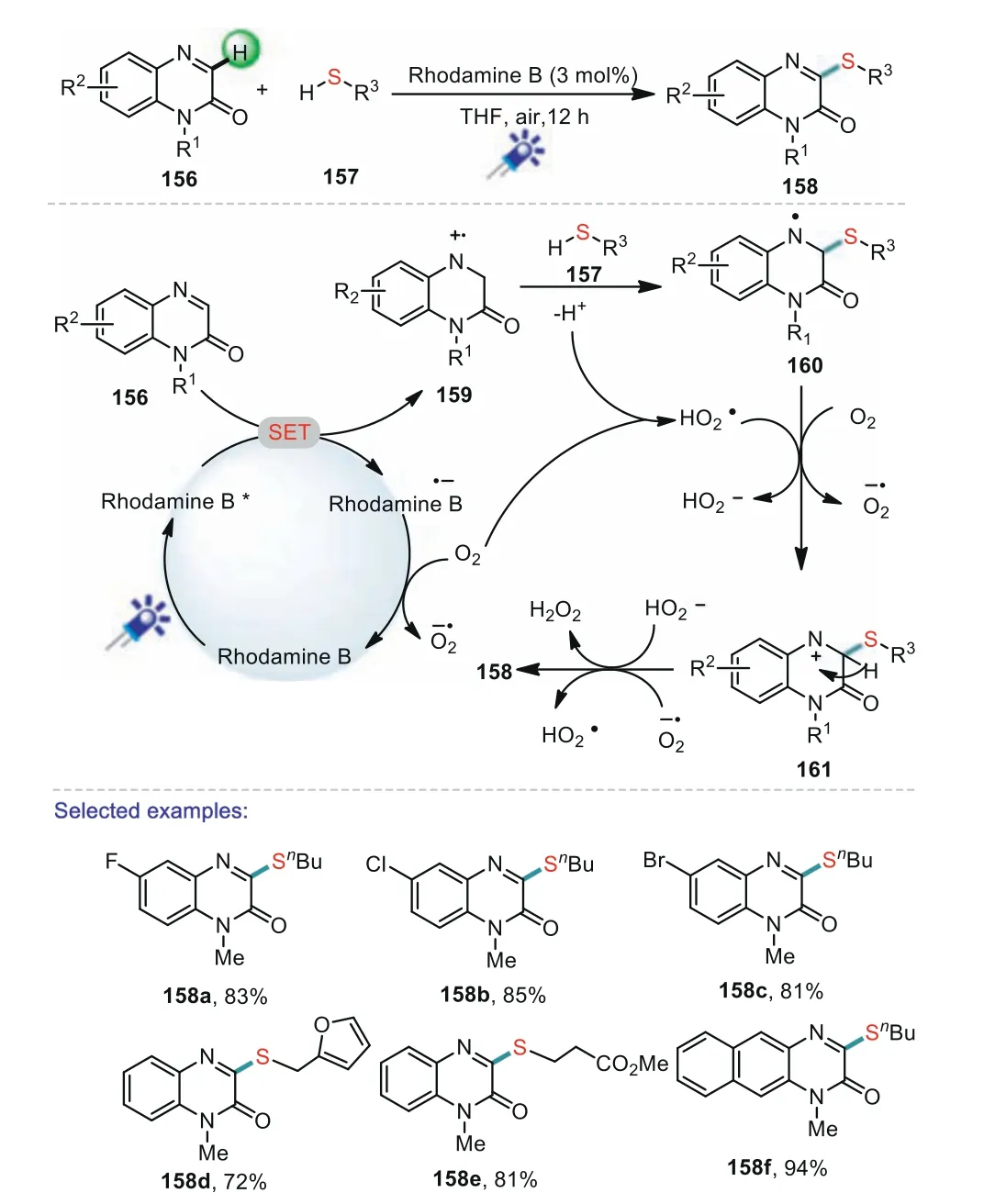
Quinoxalin-2(1H)-one derivatives have been widely applied in the areas of medicinal chemistry,materials sciences,and agrochemicals[96].Especially,3-substituted quinoxalin-2(1H)-ones are highly critical structural skeletons existing in biologically active molecules and natural products.Recently,numerous C3–H functionalization reactions of quinoxalin-2(1H)-ones have been extensively developed[97–102].In 2019,Xie and co-workers reported a new and efficient visible-light-promoted approach to 3-sulfenylated quinoxalin-2(1H)-ones through direct C–H sulfenylation of quinoxalin-2(1H)-ones with thiols under mild conditions[103].In this reaction,various 3-sulfenylated quinoxalin-2(1H)-ones can be efficiently and conveniently obtained in excellent yields with good functional group tolerance.In addition,diverse cyclic substituted thiols can participate in the reaction well,and showed a high reactivity.A possible mechanism for this reaction is depicted in Scheme 23.Initially,Rhodamine B was excited under the presence of blue LED light to produce Rhodamine B*.Then,a SET process from quinoxalin-2(1H)-one 156 to Rhodamine B*afforded the reactive radical cation 159 and Rhodamine B·-.Subsequently,the Rhodamine B·-radical anion was oxidized by O2to generate the ground state Rhodamine B and O2·-.Next,the nucleophilic addition of thiol 157 to radical cation 159 afforded nitrogen radical intermediate 160.The further oxidation of intermediate 160 by O2or HO2·gave the nitrogen cation intermediate 161.Finally,the deprotonation of 161 delivered the end product 158.
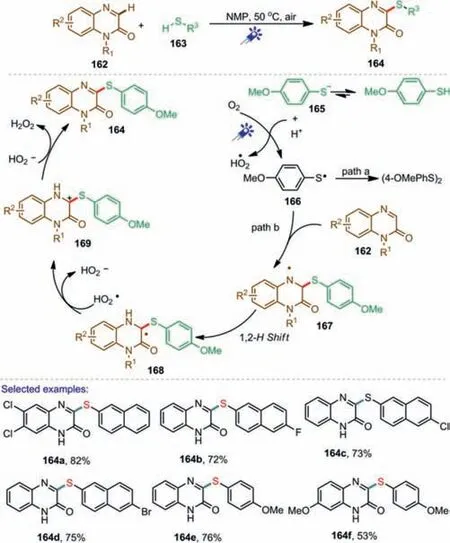
Almost at the same time,Teng and co-workers also disclosed a similar strategy of visible-light-induced sulfenylation of quinoxalinones with thiolsviacross-dehydrogenative coupling under photocatalyst-free conditions[104].In this developed method,the different thiols with either electron-withdrawing(F,Cl and Br)or electron-donating groups(OMe and Me),were all efficiently converted to the corresponding 3-sulfenylquinoxalinones in good yields.It is worth noting that for quinoxalinone containing different substituents,the groups of-OMe,-Me,and-NO2were slightly lower reactivity than those with-Cl,-Br,and-CF3.A possible catalytic cycle was proposed as shown in Scheme 24.Firstly,4-methoxybenzenethiol was transformed into thiol anion 165 and H+.Oxidation of 165 by O2in air under irradiation of a blue LED light led to thiyl radical 166.Nucleophilic radical 166 was selectively added at the electron-deficient C=N bond of 162 to generate 167.Then 167 underwent a 1,2-hydrogen shift to provide 168 which then underwent oxidation to afford carbon-centered 169.Finally,the target product 164 was generated by the deprotonation of 169.
Scheme 22.Visible-light-induced sulfenylation of imidazopyridines with thiols.
Scheme 23.Visible-light-promoted sulfenylation of quinoxalin-2(1H)-ones with thiols.
Scheme 24.Photocatalyst-free visible-light-promoted sulfenylation of quinoxalin-2(1H)-ones with thiols.
Scheme 25.Photoinduced,copper-catalyzed C-S couplings.
3.2.Photochemical sulfenylation of aryl/heteroaryl halides
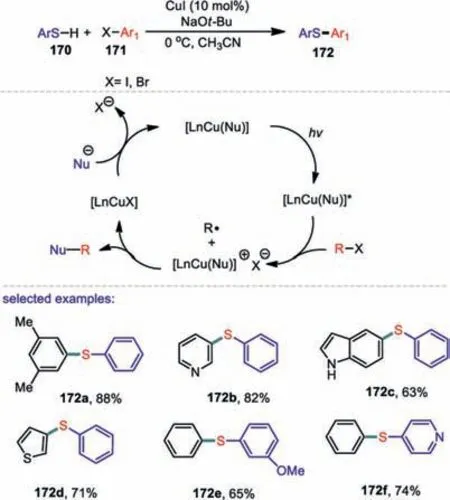
Mergingphotoredoxcatalysiswithmetalcata lyzed cross-coupling reactions that furnish carbon-carbon and carbon-heteroatom bonds are sustainable and powerful strategies for the synthesis of pharmaceuticals,fine chemicals,and materials[105–107].In 2013,Uyeda and co-workers described a versatile and mild photoinduced,copper-catalyzed method for cross-coupling of aryl thiols with aryl halides(Scheme 25)[108].The attractive advantages of this developed protocol is that the reaction can proceed under mild conditions(0 °C or below)and use inexpensive CuI as a precatalyst,and no additional ligand is necessary.As for the thiols,a various array of aryl thiols serves as suitable cross-coupling partners.Formation of the sulfenylation products was clearly explained in the catalytic cycle that these couplings might proceedviaa SET process from an excited state of a Cu(I)-nucleophile complex to the organic electrophiles.
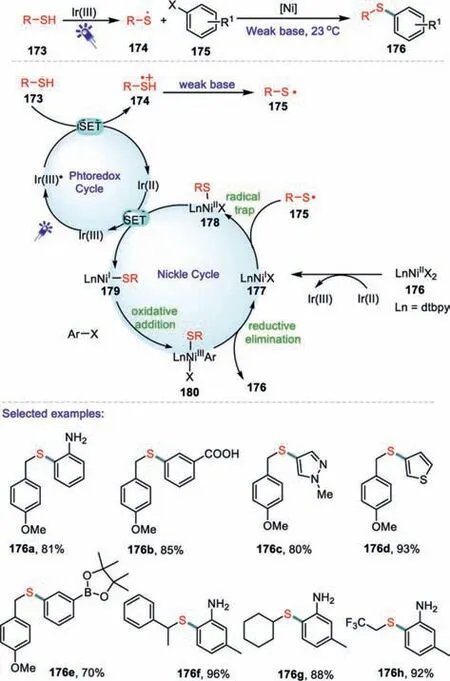
Scheme 26.Photoinduced,nickel-catalyzed C-S couplings.
In 2016,an efficient method for Ni-catalyzed cross-couplings of benzyl,aryl and alkyl thiols with aryl and heteroaryl iodides were achieved by Oderinde and co-workers(Sheme 26)[109].This C-S cross-coupling protocol is compatible with a wide range of functional group tolerance and the reactions can be carried out in the presence of O2.The developed C-S cross-coupling protocol exhibits excellent functional group tolerance and the reactions can be performed under O2atmosphere.Notably,a wide range ofortho-substituted aryl iodides bearing-F,-CH3,–NH2,and–CH3groups participated in the cross-coupling reactions to form C-S bonds in good yields.Heteroaromatic iodides,which are core units in the preparation of many bioactive compounds,such as pyrimidine,pyridine,indole,thiophene,and protected pyrazole are all effective electrophiles in this C-S cross-coupling method.A vast array of thiols are effective coupling partners,such as thiophenols and simple alkyl thiols.Based on a series of control experiments,a proposed mechanism for the dual catalysis is shown in Scheme 26.Initially,the heteroleptic Ir(III)-photocatalyst was excited under the presence of blue LED light to produce a long-lived excited state*Ir(III).Subsequently,the thiol 173 underwent a SET process to provide the thiol radical cation 174 and Ir(II).Deprotonation of the thiol radical cation 174 by pyridine generated a thiyl radical 175.Then a SET reduction of the 176 by Ir(II)delivered a Ni(I)-halide 177.Meanwhile,175 intercepted the Ni(I)-halide 177 to give a Ni(II)-species 178,which was reduced by Ir(II)to a Ni(I)-sulfide complex 179.Oxidative addition of an aryl iodide to 179 afforded a Ni(III)-complex 180,which underwent a facile reductive elimination process to form the final product 176.
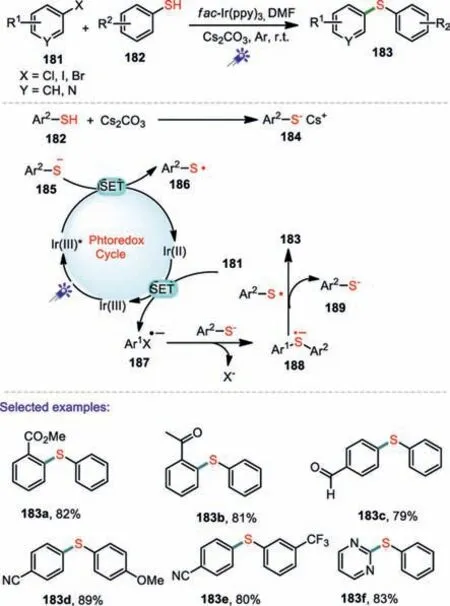
Scheme 27.Visible-light photoredox arylation of thiols with aryl halides.
Aryl chlorides are inexpensive and easy to obtain.However,their low reactivity greatly limits their wide application in transition-metal-catalyzed cross-coupling reactions.In addition,the deactivation of transition-metal catalysts by thiols has inspired organic chemists to use specially designed ligands,strong bases,and high temperatures.These factors prevent this kind of reaction from being widely used.In 2017,Jiang and co-workers reported a facile and efficient visible-light photoredox arylation of thiols with aryl halides under mild conditions(Scheme 27)[110].In the developed methods,the substrates of aryl chlorides could tolerate similar functional groups to those possible with aryl bromides and iodides.Moreover,several aryl fluorides bearing electronwithdrawing groups were tested and afforded the corresponding products in good yields.A possible mechanism for this reaction is depicted in Scheme 27.The treatment of an aryl thiol 182 with Cs2CO3afforded the corresponding thiolate 184.The photo-catalyst Ir(III)was excited under the presence of blue LED light to produce an excited state*Ir(III).The thiol anion 185 underwent a SET process by the oxidation of photoexcited*Ir(III)provided both the sulfur-centered radical 186 and Ir(II).Subsequently,the reduction of 181 by Ir(II)delivered radical anion 187 and regenerates the photocatalyst Ir(III).The 185 reacted with 187 to produces the sulfur-centered radical anion 188,and single-electron transfer from 188 to 189 forged the target product 183.
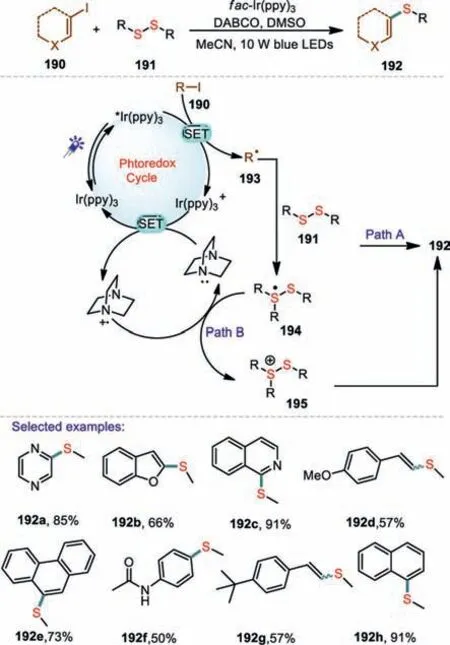
Scheme 28.Visible-light photocatalytic thiolation of aryl and vinyl iodides.
Disulfides are an attractive alternative to thiols as thiolating reagents because they are stable to air,widely available,and relatively non-volatile.In 2018,a complementary,general approach to aryl and vinyl alkyl thioethers from a range of aryl,heteroaryl and vinyl iodides has been reported by Brzozowski and co-workers(Scheme 28)[111].A wide range of thioether products containing various functional groups can be obtained in high yield as well as with excellent chemoselectivity.It is worth noting that the developed method is compatible with oxygen,sulfur and nitrogen containing heterocycles,highlighting the applicability of this method to the construction of biologically important skeletons.On the basis of the control experiments,they proposed the following mechanism for the C–S bond forming reaction.Initially,the iodide 190 underwent a SET process from[Ir(ppy)3]*to provide an[Ir(ppy)3]+and aryl radical 193.The aryl radical 193 was quenched with dialkyl disulfide 191 to produce a sulfur radical adduct 194.Then,195 underwent direct homolysis of sulfur-based radical to form thioether products 192.On the other hand,DABCO could function as an electron shuttle and the possibility of it acting as an oxidant for the sulfur radical 194 and produce thioether products 192.
With the energy crisis and the growing demand for environmental protection,the development of green and metal-free synthetic strategies is greatly desired.In 2017,Liuet al.develoed a mild and facile visible-light-induced cross-coupling reaction between thiols and arylhalides for the construction of C-S bonds without either transition metal or photoredox catalysts(Scheme 29)[112].In this developed method,a various of thiols containing electron-donating and electron-withdrawing groups were successfully coupled with aryl halides to yield products in good to excellent yields.In addition,this metal-free protocol for C-S coupling reaction was used in late-stage modifications of active pharmaceutical molecules and drug synthesis.DFT calculations and UV-vis spectroscopy indicates that visible-light induced intermolecular charge transfer within the thiolate-aryl halide electron donor-acceptor complex promoted the reactivity in the absence of a transition metal or a photocatalyst.
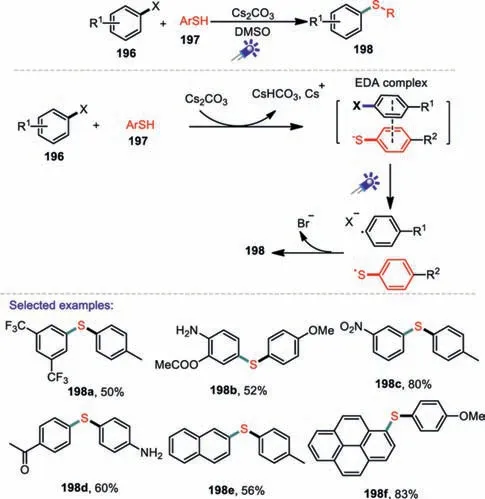
Scheme 29.Visible-light-promoted C-S formation via intermolecular charge transfer.
S-Aryl dithiocarbamates are ubiquitous in a variety of biologically active compounds and natural products.In 2019,Li and coworkers successfully developed a green and efficient multicomponent reaction protocol to synthesizeS-aryl dithiocarbamates under visible light conditions[113].The reaction can proceed smoothly without adding any transition-metal catalysts,ligands,or photocatalysts while minimizing chemical wastes and metal residues in the end products.Notably,aryl iodides bearing the electronwithdrawing groups displayed high reactivity.It is worth noting that aromatic amines showed good reactivity compared to alkyl amines.Satisfactorily,this transformation can be applied to the late-stage functionalization of a pharmaceutically relevant molecule.A possible mechanism for this reaction is shown in Scheme 30.Initially,CS2would react with amines in the presence of Cs2CO3to give the corresponding thiolate 203.Then,an aryl halide and a thiolate anion first associated,forming an EDA complex.This EDA complex was subsequently activated by visiblelight irradiation to induce a SET from the thiolate anion to the aryl halide to generate a thiyl radical,a halide anion,and an aryl radical 204.Finally,the aryl radical coupled with the thiyl radical,forming the coupling product 202.
3.3.Photochemical sulfenylation of aryl/heteroaryl amine derivatives
The cleavage of C-N bond also provides an effective approach to obtain carbon-centered free radicals.In the past few decades,nitrogen-containing precursors with relatively weak C-N bonds,such as aryl diazonium salts,aryl hydrazide,have been successfully applied to the generation of various carbon-centered radicals[114–116].In 2017,Hong and co-workers reported a mild and convenient visible light photocatalysis method for the synthesis of diaryl sulfides in the presence of Eosin Y[117].As for diazonium salts,ortho and meta-substituted aryl diazonium salts were welltolerated in the present transformation.Electron-deficient aryl diazonium salts also forged the products in high yields.Two plausible reaction pathways are shown in Scheme 31.First,the irradiation of eosinY(EY)with blue visible light generated its excited state species EY*that could interacted with O2to provide singlet oxygen(1O2).Then,the singlet oxygen(1O2)obtained a hydrogen atom from thiol to afford thiyl radical 209,which reacted with aryl radical 210 generated from aryl diazonium salt 206 to produce the desired diaryl sulfide 207(Path A).On the other hand,the thiyl radical 209 could also be produced through a one-electron oxidation process of thiol(Path B).The thiol radical cation 208 by the Eosin Y radical cation could react with O2·-to give thiyl radical 209 for further reaction with aryl radical 210 to generate the final product 207.Under the similar conditions.In 2019,Liet al.reported another photocatalytic method for the construction C-S bond using cercosporin as a photocatalyst(Scheme 32)[118].
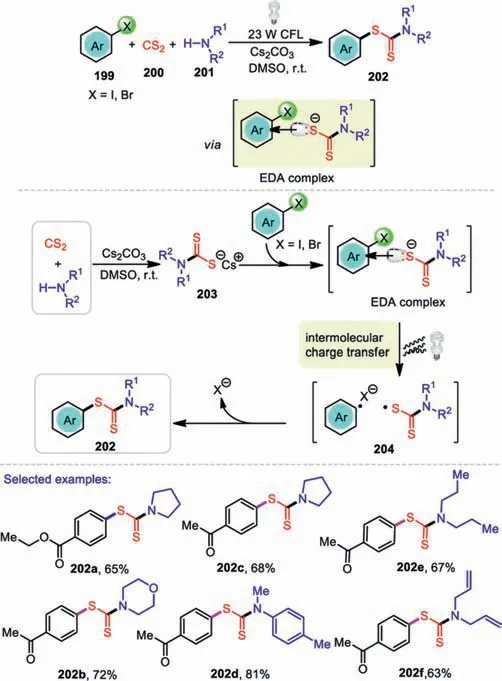
Scheme 30.Visible-light-promoted C-S coupling for the formation of S-aryl dithiocarbamates.
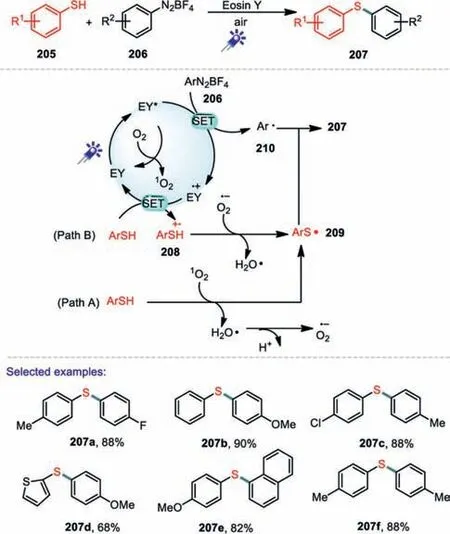
Scheme 31.Visible-light-promoted C-S coupling from aryl thiols and aryl diazonium salts.
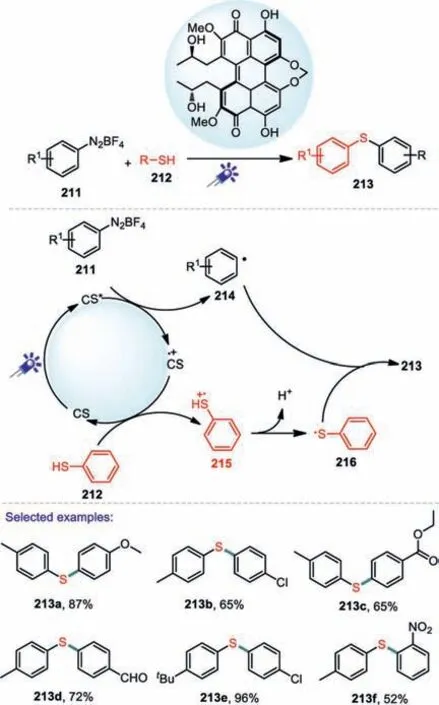
Scheme 32.Cercosporin-Photocatalyzed C-S Coupling for the formation of C-S bond.
Recently,arylhydrazines have attracted considerable attention as alternative electrophilic partners in cross coupling reactions.In 2019,Li and co-workers presented an efficient visible-light-induced sulfidation of arylhydrazines for the synthesis of aromatic sulfides(Scheme 33)[119].The heterocycle containing hydrazine,reacting with diaryl disulfides also gave the desired product in good yield.It should be noted that diaryl disulfides bearing electron-donating groups,such as–OMe and–NH2,were successfully afforded the desired products effectively.However,diaryl disulfides bearing a strong electron-withdrawing group showed relatively poor reactivity.A plausible mechanism was proposed.Initially,under the irradiation of blue visible light,PC(Na2Eosin Y)was excited to the photo-excited state of PC*,which was then converted into PC·-by a SET process.Subsequently,the oxidation of PC·-by O2formed the ground state photocatalyst and O2·-,while concomitantly,phenyl hydrazine 217 was oxidized to provide radical cation 220.The deprotonation of radical cation 220 by O2·-or base gave radical 221.Thenviaa SET process followed by deprotonation generated 221 which was converted into 222,and then into radical 223.Subsequently,the elimination of nitrogen from 223 generates the phenyl radical 224.On the other hand,the phenyl sulfide radical 225 was obtained through homolytic cleavage of 218 under visible-light irradiation.Finally,the radical coupling of 224 and 225 delivered the corresponding product diphenyl sulfide 219.
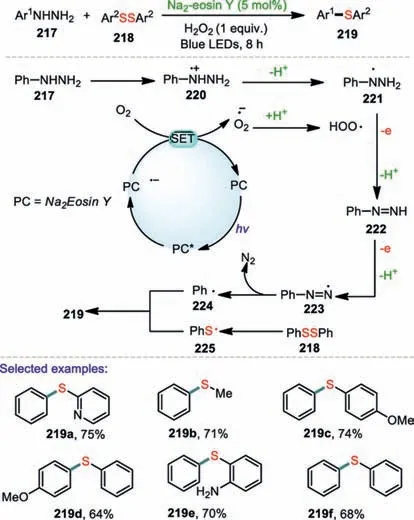
Scheme 33.Visible-light-promoted sulfidationof arylhydrazines.
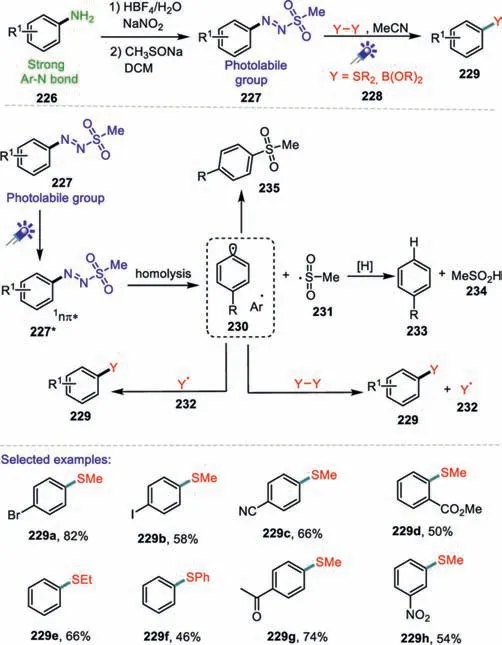
Scheme 34.Visible-light-promoted C-S bond formation using arylazo sulfones.
Recently,arylazo sulfones have been successfully used in the metal-free arylation of heterocycles and unactivated arenes without the need of additives[120,121].In 2019,Blank and co-workers reported an elegant photoinduced synthesis of aryl sulfides under metal-and photocatalyst-free conditions using arylazo sulfones as the arylating reagent(Scheme 34)[122].The current protocol is a sustainable method that uses a bench-stable arylazo sulfones,which is easily prepared from anilines,and allows wavelength selective generation of aryl cations and aryl radicals.Diethyl sulfide and diphenyl sulfide were also successful in the photocatalyst-free thioether synthesis resulting in the final products in 50% and 46%yield,respectively.A plausible mechanism was proposed as shown in Scheme 34.The irradiation of 227 with blue visible light generated the photo-excited state1nπ*-state 227*.Subsequent homolytic cleavage of the S–N bond delivered the aryl radical 230 and methanesulfonyl radical 231.Such radicals were trapped by the disulfide reagent 228 to afford the products 229 and the sulfide radical(Y·)232.Then aryl combined with Y-radicals to provide 232.Hydrogen atom abstraction by the generated radicals from the solvent to give arenes 233 and sulfinic acid 234.Finally,the recombination of the aryl 230 and methanesulfonyl radical 231 resulted in a second possible by-product sulfone 235.
3.4.Photochemical cyclization reaction for C(sp2)-S bond formation
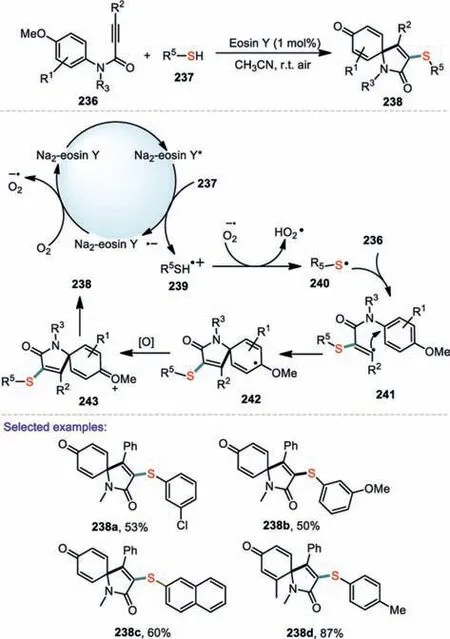
Cascade cyclization reaction is a powerful organic transformations for the construction of carbocyclic and heterocyclic skeletons from relatively simple acyclic motifs[123].In recent years,the difunctionalization of alkynesviaelectrophilicipso-cyclization or cascade radicalipso-cyclization has been proved to be a highly attractive and efficient protocol for the synthesis of various functionalized azaspiro[4,5]trienones[124–126].In 2017,Wei and coworkers reported a convenient and efficient visible-light-promoted method for the construction of 3-sulfenyl azaspiro[4,5]trienones through metal-free difunctionalization of alkynes with thiols at room temperature[127].This method uses visible light as the safe and eco-friendly energy source,and inexpensive and nontoxic organic dyes as photo-catalysts affording various sulfur-containing azaspiro[4,5]trienones in moderate to good yields.As for the arylalkynyl,with either electron-withdrawing or electron-donating groups,were all efficiently converted to the corresponding products.It is worth nothing that the amide with a N–H group was also successful to afford the desired product in the present reaction system.Based on the experiments results and previous reports,a possible reaction mechanism was described in Scheme 35.Firstly,The irradiation of Na2-Eosin Y with blue visible light generated the photo-excited state of Na2-Eosin Y*.Subsequently,Na2-Eosin Y*got a single electron from 237 to generate the radical cation 239 and Na2-Eosin Y·-radical anion.Then Na2-Eosin Y·-underwent oxidation of O2(air)to provide the ground state Na2-Eosin Y and O2·-.The radical cation 239 was deprotonated by O2·-leading to the thiol radical 240.The resulting thiol radical reacted with 236 to deliver the vinyl radical 241.Next,the intramolecular spirocyclization of the vinyl radical with an aryl ring generated the radical intermediate 242.Finally,242 was oxidized to afford the desired oxygenium intermediate 243,which was converted into the final 3-sulfenyl azaspiro[4,5]trienone 238.
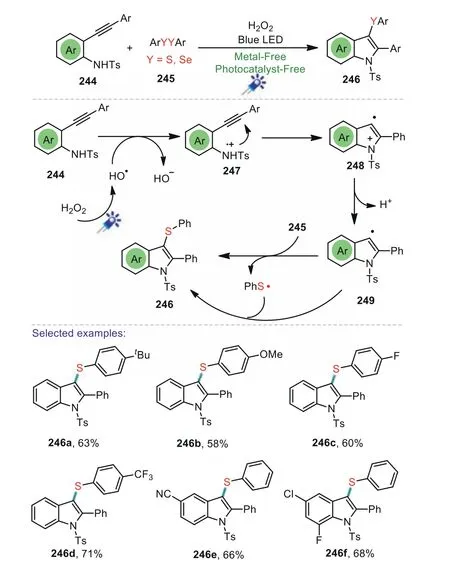
The indole framework is a key core scaffold exists widely in natural products,and biological active molecules[128].Recently,the visible light-induced formation of the indole skeleton has attracted considerable attention in the synthetic community.In 2017,Shi and co-workers demonstrated an efficient and convenient strategy for the construction of 3-sulfenyl indolesviaa visible-lightpromoted domino cyclization of 2-alkynylanilines with disulfides(Scheme 36)[129].The reaction was induced by visible-light irradiation in the presence of H2O2under transition metal-and photocatalyst-free conditions to generate the corresponding products in good yields.Control experiments indicated that a free radical was involved in the present transformation.Mechanistically,treatment of 2-alkynylaniline 244 with hydroxyl radical,which was produced from the homolytic cleavage of H2O2under blue LED irradiation,to give an intermediate 247.Then,the obtained 247 underwent intermolecular cyclization to deliver intermediate 248,followed by a deprotonation to generate intermediate 249.Finally,249 interacted with diphenyl disulfide 245 to provide the end product 3-sulfenylindole 246.
Scheme 35.Visible-light-promoted approach to 3-sulfenyl azaspiro[4,5]trienones.
Scheme 36.Visible-light-induced tandem oxidative cyclization of 2-alkynylanilines with disulfides.
Scheme 37.Visible-light-induced charge-transfer radical spirocyclisation.
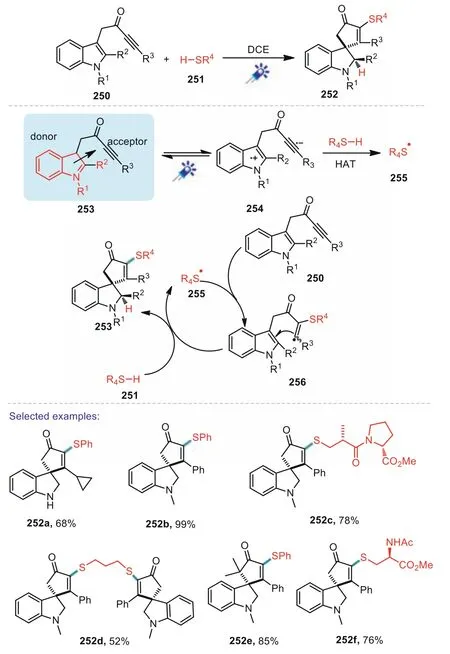
In 2020,Unsworth reported an elegant visible-light-promoted intramolecular charge transfer in the radical spirocyclisation of indoletethered ynones(Scheme 37)[130].A wide range of sulfurcontaining spirocycles have been constructed using this high yielding and mild synthetic strategy.The key of this process is the formation of an intermolecular EDA complex between an indole(the donor)and a pyridinium salt(the acceptor);the EDA complex can then absorb visible light to promote charge transfer.Substituted aryl thiols of various electronic effects were well tolerated in the methods developed.Gratifyingly,aliphatic thiols are also compatible with the standard conditions.A mechanism was proposed that after the formation of EDA complex 253,a photoexcited state was generated through visible light absorption,which was loosely represented as charge transfer complex 254.This species might simply relax to reform EDA complex 253viaback electron transfer,or alternatively,the thiol gave a hydrogen atom to the excited state 254 to afford 255 which was needed to start a radical cascade.Finally,thiyl radical reacted with the ynone to afford the final products 252(Scheme 37).
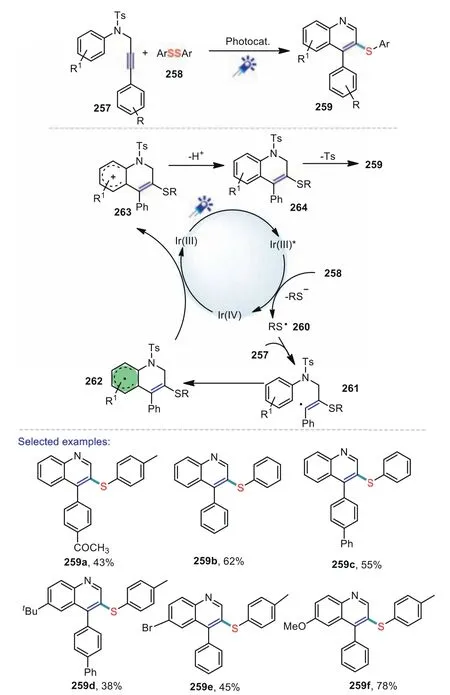
Scheme38.Visible-lightinducedtandemcyclizationofN-aryl-Ntosylpropargylamine with disulfides.
Quinoline and its derivatives represent an important class of naturally occurring heterocycles for its efficient biological and physiological activities[131].Studies for constructing 3-substituted quinolines are still an important goal in the field of organic chemistry.In 2019,Wanget al.reported an efficient one-pot method for the synthesis of 3-phenylthioquinolinesviavisible-light promoted cyclization ofN-aryl-N-tosylpropargylamine with disulfides[132].This unprecedented method,involving C-S bond formation,detosylation and aromatization,provided the 3-phenylthioquinolines in a wide range substrate scope under mild conditions.According to the results of the control experiments,a possible reaction pathway was proposed in Scheme 38.Firstly,the photoredox catalystfac-Ir(ppy)3was irradiated to the excited state Ir*(III),which underwent oxidative quenching with 258 to afford radical 260 and Ir(IV).Then radical 260 underwent intermolecular addition to 257 to afford radical 261.Subsequently,the radical 261 went through intramolecular cyclization to generate radical 262 and then it was oxidized to produce carbocation 263 by Ir(IV).Carbocation 263 underwent deprotonation to give compound 264 which subsequently experienced detosylation and aromatization to provide the final product 259.
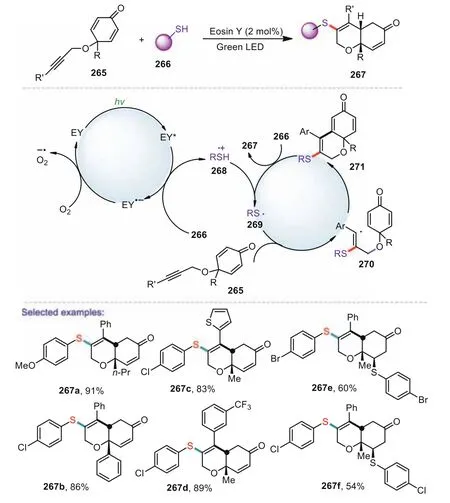
Scheme 39.Visible light mediated thiol–yne/conjugate addition cascade of alkynetethered cyclohexadienones.
Organic dyes are increasingly utilized as an attractive alternative to the transition-metal complexes in photoredox catalysisdue to their advantages of being inexpensive,easily available and less toxic[133].In 2019,Nair and co-workers reported an elegant approach to sulfenylated dihydrochromenones through visible-lightinduced thiol–yne/conjugate addition cascade of alkyne-tethered cyclohexadienones(Scheme 39)[134].The reaction shows good functional group tolerance and is carried out in metal-free conditions using cheap and readily available Eosin Y as the photocatalyst and using green light as a sustainable energy source.Notably,aliphatic thiols are as effective as the aromatic ones giving excellent yields of the cyclized products.Gratifyingly,sensitive functional groups like nitro,acetyl and trifluoromethyl are compatible under the reaction conditions.A plausible mechanism is shown in Scheme 39.Under the irradiation of Eosin Y with green visible light generated the photo-excited state of Eosin Y*,which on reductive quenching by thiol 266 generated Eosin Y·-and thiyl radical cation 268.The radical cation 268 lost a proton to form the thiyl radical 269,which was added to alkynes to give a vinyl radical 270 which was stabilized by an aryl ring.The intermolecular cyclization of the radical to give the intermediate 271.Then the intermediate 271 abstracted a hydrogen atom from another molecule of thiol and generate the end product 267 and thiyl radical 269.Finally,Eosin Y·-was oxidized by an O2to regenerate the active photocatalyst Eosin Y.
4.Conclusions and outlook
Over the past few years,extensive experimental work in visiblelight catalyzed cross-coupling and cyclization reactions has resulted in significant advances for C–S bond formation under mild conditions.The progress summarized in this review highlights the photochemical C(sp3)-S and C(sp2)-S bond formation that have been developed in organic synthesis.In these processes,the discovery and development of simple and efficient photocatalytic systems and rational design of substrate types play an important role.As shown in the above mentioned examples,this strategy is quite valuable for the construction of diverse sulfur-containing molecules.
Despite great progress has been made,however,many opportunities and challenges still remain as follows:(1)In terms of the visible-light-promoted C(sp3)-S bond construction,the substrate scope is limited.Therefore,the development of new photocatalytic systems to achieve the diversity of substrates activation and transformation is still a large research space;(2)Photocatalystfree visible-light-promoted sulfenylation of arylhalides has been developed,however,the substrates are mainly focus on aryl halides with electron-withdrawing groups;(3)The visible-light-induced C-S bond construction reaction directly using sulfur powder as the sulfur source has not been achieved thus far;(4)Visible-lightpromoted C(sp)-S bond formation has not been achieved thus far,so it is highly desirable to develop new catalytic systems to achieve this transformation.All of these issues need further exploration,and the results need further improvement.Visible-light-catalyzed C-S bond formation is still a vigorous research area with both great prospect and huge challenges.We believe there will be more and more innovative achievements presented in the near future.
Declaration of competing interest
The authors declare that they have no known competing financialinterestsor personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the Natural Science Foundation of Shandong Province(No.ZR2016JL012),Hunan Provincial Natural Science Foundation of China(No.2019JJ20008),and the Scientific Research Foundation of Qingdao University of Science and Technology(No.1203043003457).
 Chinese Chemical Letters2022年4期
Chinese Chemical Letters2022年4期
- Chinese Chemical Letters的其它文章
- Key progresses of MOE key laboratory of macromolecular synthesis and functionalization in 2020
- Small nanoparticles bring big prospect:The synthesis,modification,photoluminescence and sensing applications of carbon dots
- Cell membrane-coated nanoparticles for immunotherapy
- Diketopyrrolopyrrole-derived organic small molecular dyes for tumor phototheranostics
- Exosome based miRNA delivery strategy for disease treatment
- Recent advances in targeted stimuli-responsive nano-based drug delivery systems combating atherosclerosis