
Design and synthesis of novel α-aminoamides derivatives as Nav1.7 inhibitors for antinociception
2022-06-18 10:53DengqiXueYniLiuYilinZhengHelingNiuLiyingDongXingshuoOuyngSiyuSongDenggoZhngQinweiGeKeweiWngLimingSho
Chinese Chemical Letters 2022年3期
Dengqi Xue,Yni Liu,Yilin Zheng,Heling Niu,Liying Dong,Xingshuo Ouyng,Siyu Song,Denggo Zhng,Qinwei Ge,Kewei Wng,Liming Sho,b,∗
a School of Pharmacy,Fudan University,Shanghai 201203,China
b State Key Laboratory of Medical Neurobiology,Fudan University,Shanghai 200032,China
c Department of Pharmacology,School of Pharmacy,Qingdao University Medical College,Qingdao 266071,China
Keywords:α-Aminoamides Sodium channel Nav1.7 inhibitor Chronic pain Analgesia
ABSTRACT Three novel series of α-aminoamides derivatives were designed and synthesized based on ralfinamide,and their Nav1.7 inhibitory activities were evaluated using manual patch clamp electrophysiology.Active compounds inhibited Nav1.7 with half maximal inhibitory concentration (IC50) values ranging from 2.9 μmol/L to 21.4 μmol/L.Among them,the most potent compound 19h exhibited about 12-fold potency better than ralfinamide.The investigation of their structure-activity relationship gives a strategy to improve the Nav1.7 inhibition of ralfinamide analogues.Compound 19h was efficacious in antinociception in the mouse spared nerve injury (SNI) model of neuropathic pain without causing sedation in the open field test.
Chronic pain has serious and negative impact on quality of life[1-3].Currently available analgesics are limited with either greater side-effects or low levels of analgesic efficacy,thus presenting an unmet medical need [4].Voltage-gated sodium channel Nav1.7 encoded bySCN9Agene plays a key role in transmission of pain signaling [5,6].The loss-of-function mutations in Nav1.7 lead to congenital insensitivity to pain (CIP) [7,8].Conversely,the gainof-function mutations in Nav1.7 underlie inherited erythromelalgia,paroxysmal extreme pain disorder and idiopathic small fiber neuropathies [5,9].These lines of evidence indicate that Nav1.7 is a promising target for pain therapy.Ralfinamide (Fig.1),a Nav1.7 inhibitor,was developed by Newron Pharmaceuticals,it also shows inhibition toward to Cav2.2 andN-methyl-D-aspartate (NMDA) receptor [10-14].Ralfinamide was under phase III clinical trial for low back pain before discontinued [15].Nav1.7 is a promising target for pain therapy,so optimizing the structure of ralfinamide may lead to a compound with higher Nav1.7 inhibition and betterin vivoefficacy in antinociception.
In this study,three series of novelα-aminoamides derivatives were designed and synthesized for investigation of their structure–activity relationship (SAR).Started with investigation of the influence of the length of carbon linkage between theα-amino group and the aryl group (Fig.1,I),then,we modified theα-aminoamide groups (Fig.1,II) before various substituents were introduced into the benzyl group for investigation of the SAR (Fig.1,III).Seventeen novelα-aminoamides derivatives were obtained and their inhibitions of human Nav1.7 currents were tested in whole-cell patch clamp recording assay.A lead compound was further evaluated for itsin vitrometabolic stability andin vivoantinociception.
The syntheses ofα-aminoamides derivatives were depicted in Scheme 1.Detailed procedures and compound characterizations can be found in Supporting information.4-Hydroxyphenethyl alcohol as a starting material reacted with 2-fluorobenzyl bromide in acetone to give compound 2 and then it was oxidized by the Dess-Martin periodinane to yield the compound 3 [16,17].Compound 3 reacted further with L-alaninamide hydrochloride in the presence of sodium cyanoborohydride to yield the target compound 4.The compound 6 was achieved by the reaction of methyl 3-(4-hydroxyphenyl) propionate with 2-fluorobenzyl bromide in acetonitrile,subsequently through the hydrolysis of carboxylic ester to produce the compound 7.Compound 7 was reduced with lithium aluminum hydride to the alcohol 8,which was oxidized by Dess-Martin periodinane to yield the compound 9.The target compound 10 was obtained by the reaction of compound 9 with Lalaninamide hydrochloride.4-(4-Hydroxyphenyl)butanoic acid was refluxed with 2-fluorobenzyl bromide in acetone to afford the compound 12.Compound 12 was reduced with lithium aluminum hydride and then it was oxidized by the Dess-Martin periodinane to yield the compound 14.The compound 14 reacted further with L-alaninamide hydrochloride to give the target compound 15.The target compound 16 was achieved by the reaction of compound 10 with formaldehyde.The compound 9 reacted with appropriateα-aminoamides hydrochloride to yield the target compounds 17a–d.The compound 18 was obtained by removing theortho-fluorobenzyl group from compound 10,which reacted further with a series of substituted benzyl alcohol to yield the target compounds 19a–i.
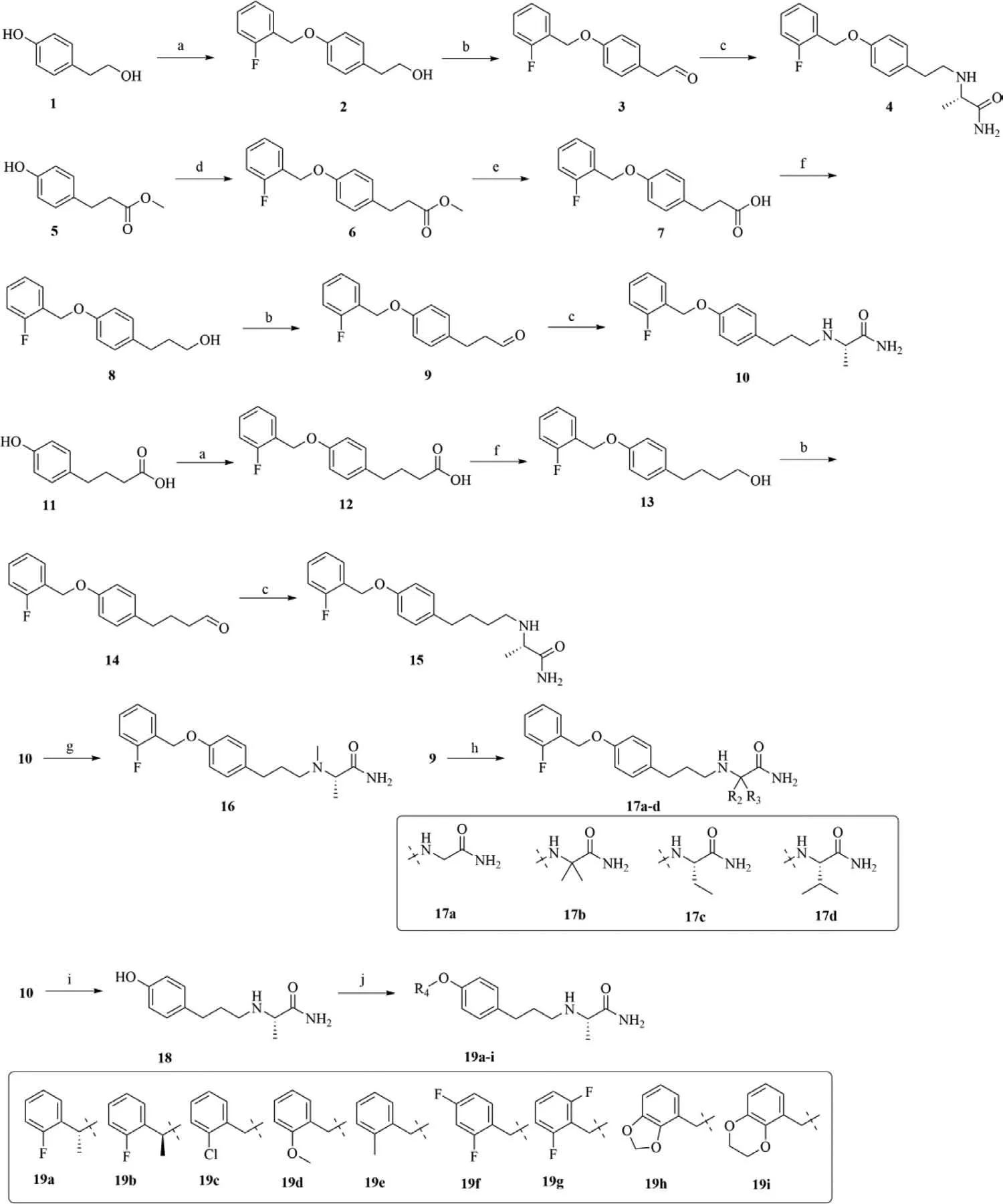
Scheme 1.Synthesis of α-aminoamides derivatives.Reaction conditions: (a) 2-Fluorobenzyl bromide,K2CO3,acetone,reflux,63%–96%.(b) Dess-Martin periodinane,CH2Cl2,r.t.,43%–88%.(c) L-Alaninamide hydrochloride,NaBH3CN,MeOH,AcOH,r.t.to 60 °C,43%–69%.(d) 2-Fluorobenzyl bromide,K2CO3,acetonitrile,90 °C,70%.(e) NaOH,MeOH,THF,60 °C,93%.(f) LiAlH4,THF,80 °C,60%.(g) 37% Formaldehyde (aq),NaBH3CN,MeOH,AcOH,r.t.,88%.(h) Appropriate α-aminoamides hydrochloride,NaBH3CN,MeOH,AcOH,r.t.,44%–71%.(i) Hydrogen,5% palladium on activated carbon,EtOH,45 °C,90%.(j) Substituted benzyl alcohol derivatives,PPh3,di-tert-butyl azodicarboxylate (DBAD),THF,90 °C,18%–57%.
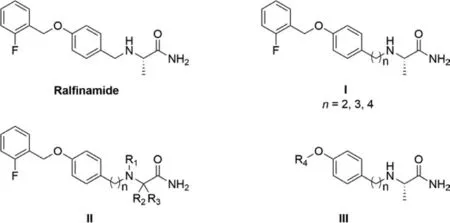
Fig.1.Chemical structures of ralfinamide and its designed compounds (I,II and III).
All synthesized compounds (4,10,15,16,17a–d and 19a–i)were tested for their inhibition of human Nav1.7 channel currents using whole-cell patch clamp recordings technique (Supporting information) [18,19].Ralfinamide was selected as reference compound.As summarized in Table 1 and Fig.2,all compounds exhibited a dose-dependent inhibition of Nav1.7 currents with half maximal inhibitory concentration (IC50) values ranging from 2.9 μmol/L to 21.4 μmol/L.Among them,the compound 19h (IC50=2.9 μmol/L) was the most potent with about 12-fold more potency than ralfinamide (IC50=35.2 μmol/L).The compounds 4,10 and 15 showed better potency than ralfinamide,indicating that increasing the carbon chain length between theα-amino and the aryl group was beneficial for the bioactivity.Among the three compounds,the compound 10 (IC50=5.0 μmol/L) with ideal three carbon atoms of the chain length showed the most potent inhibition of Nav1.7 current.The compound 16 demonstrated weaker inhibition than the compound 10,indicating that the methyl substitution on theαamino group was not beneficial for the bioactivity.Removing the methyl group fromα-position of aminoamides (compound 17a)or adding methyl group toα-position of aminoamides (compound 17b) reduced inhibitory activity,it also showed a steric effect of the substituents atα-position of aminoamides with a potency order of CH3(compound 10)>CH2CH3(compound 17c)>CH(CH3)2(compound 17d).Introduction of a methyl group to theorthrofluoro benzyl position (compounds 19a and 19b) led to a decrease potency with a little chirality effect.2-Cl (compound 19c),2-OCH3(compound 19d) and 2-CH3(compound 19e) substitution on the phenyl ring led to a decreased efficiency.Alternatively,extra fluorine in either thepara- or theorthro-position (compounds 19f and 19g) showed no improvement on Nav1.7 inhibition.Whereas substituents such as 1,2-methylenedioxy (compound 19h) showed the best potency with IC50=2.9 μmol/L,which is better than a bulkier substituted group such as 1,2-ethylenedioxy (compound 19i).
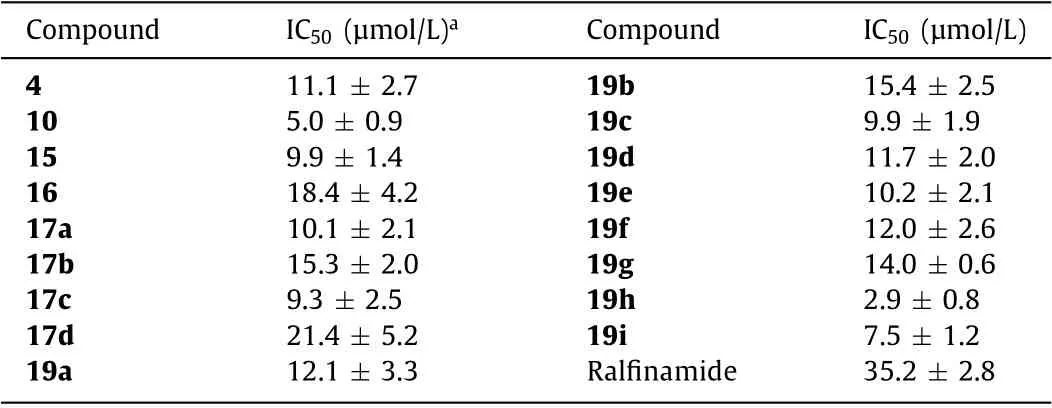
Table 1 Inhibition of Nav1.7 channel currents by α-aminoamides derivatives in whole-cell patch clamp assay.
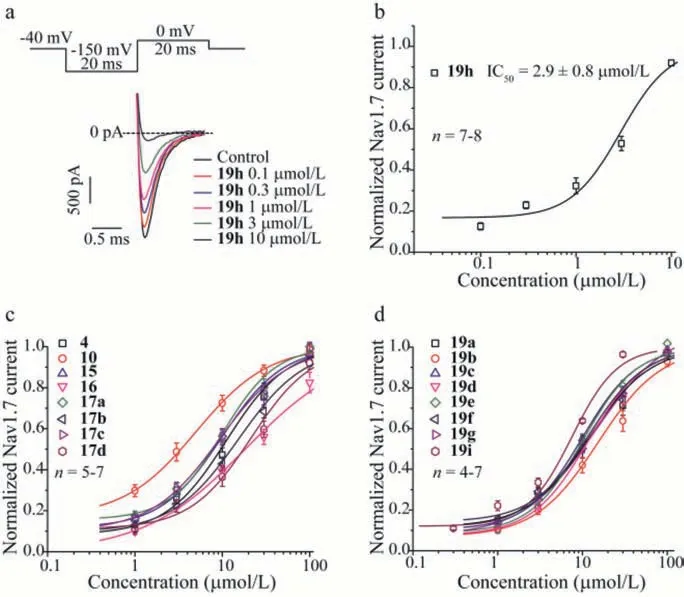
Fig.2.The dose-dependent inhibition of Nav1.7 currents by α-aminoamides derivatives in whole-cell patch clamp recordings of HEK293 cells stably expressed Nav1.7 channel.(a) Top panel,voltage protocol used in compound inhibition experiments.To activate Nav1.7 from a holding voltage of −40 mV,a 20 msec prepulse to −150 mV is applied to recover the current from inactivation followed by a 0 mV pulse to open the channel with 1 s intervals.Bottom panel,representative current traces elicited by 0 mV before and after applications of compound 19h.The dashed line represents zero current level.(b) Dose-response curve of compound 19h was fitted using logistic function with IC50 of 2.9 ± 0.8 μmol/L,n=7–8.(c) and (d) Curves were fitted using logistic function for concentration-dependent inhibition of Nav1.7 by α-aminoamides derivatives,n=4–8.All data were expressed as the mean ±SEM.

Table 2 In vitro metabolic half-life and predicted intrinsic clearance (CLint) of compound 19h in human and mouse liver microsomes.
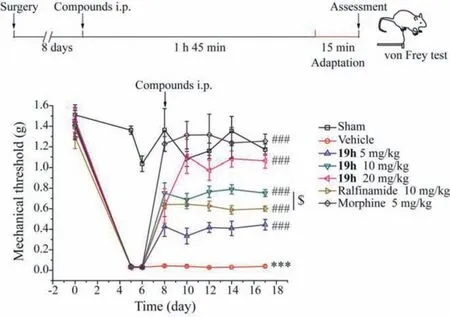
Fig.3.The dose-dependent attenuation of nociception by compound 19h in SNIinduced mechanical allodynia in mice.Top panel,a schematic time-course of paw mechanical withdrawal threshold (PMWT) measured by von Frey hair stimulation in mouse model of SNI.Bottom panel,a dose-dependent antinociception by 19h(5,10 and 20 mg/kg,i.p.),morphine (5 mg/kg) and ralfinamide (10 mg/kg) in SNI model.Two-way ANOVA followed by Bonferroni post hoc tests revealed a significant decrease of PMWT between group sham and group vehicle,∗∗∗P < 0.001;a significant increase of PMWT in 19h (5,10 and 20 mg/kg,i.p.) injected groups,compared with group vehicle,###P < 0.001;and also a significant difference between group 19h (10 mg/kg) and group ralfinamide (10 mg/kg),$P < 0.05.Data were expressed as the mean ± SEM and ∗∗∗P < 0.001 (vs.Sham),n=5–9.
It is important to study the metabolic stability of compound,because the most of the drugs are metabolized by cytochrome P450 (CYP450) in the liver [20-23].The metabolic stability of the compound 19h was tested in human and mouse liver microsomes(Table 2 and Supporting information),and 19h appeared less stable than ralfinamide likely due to its higher clearance.
The spared nerve injury (SNI) model of neuropathic pain is characterized with a significant and continuous increase in mechanical sensitivity of mice [24].Detailed procedures can be found in supporting information.The withdrawal threshold of the ipsilateral paw after surgery (0.03 ± 0.002 g) was significantly lower than the sham operation group (1.03 ± 0.07 g) on the sixth day after operation,indicating a successful establishment of mouse SNI model (Fig.3).After intraperitoneal administrations of compound 19h (5,10 and 20 mg/kg) for 10 days,the average paw withdrawal threshold was dose-dependently increased to 0.44 ± 0.05 g,0.75± 0.03 g and 1.06 ± 0.07 g,respectively,as compared with the value of 0.04 ± 0.001 g for vehicle control or 0.60 ± 0.03 g for ralfinamide at 10 mg/kg.In addition,the paw withdrawal threshold for the group with compound 19h at 20 mg/kg (1.06 ± 0.07 g)was comparable to the sham operation group (1.17 ± 0.05 g) and morphine (1.25 ± 0.07 g) at 5 mg/kg.
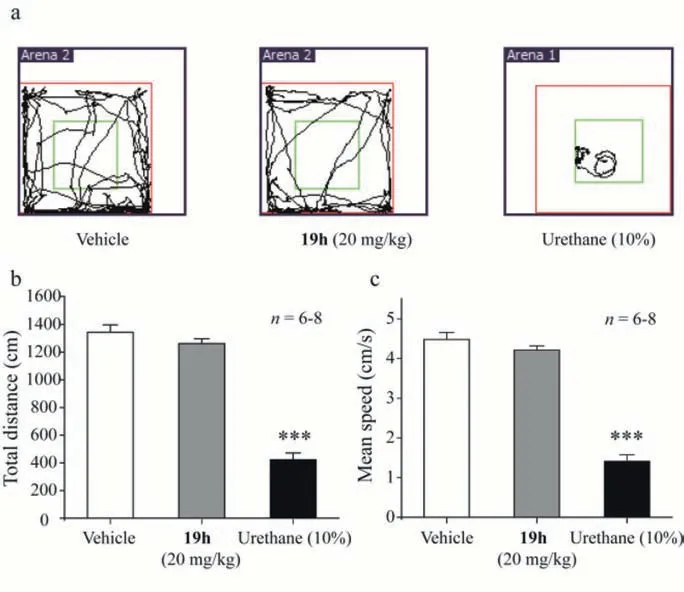
Fig.4.Compound 19h lack of altering locomotion.(a) Representative traces of mouse travels in the open field test after injections of saline,compound 19h (20 mg/kg,i.p.) and urethane (10%,i.p.).(b) The total distance traveled in the open field for 5 min after injections of saline,compound 19h (20 mg/kg,i.p.) and urethane(10%,i.p.).(c) The mean travel speed in open field for 5 min after injection of Saline,compound 19h (20 mg/kg,i.p.) and urethane (10%,i.p.).One-way ANOVA followed by Tukey test,n=6–8.All data were expressed as the mean ± SEM.
Locomotor activity is commonly used for evaluation of psychostimulative or sedative effects.We next tested the effect of compound 19h on locomotion by assessing the total travel distance and average speed in the open field test (Supporting information).As shown in Fig.4,mice treated with 20 mg/kg compound 19h (i.p.)had no significant differences in the total travel distance and the average speed,as compared with the vehicle control group.This result suggests that the compound 19h induced-antinociception was less likely resulted from the sedation effect.
With ralfinamide as a starting point,we synthesized a variety ofα-aminoamides derivatives that were evaluated against Nav1.7 channel using whole-cell patch clamp recordings.Among the tested compounds,the compound 19h was the most potent with about 12-fold more potency than ralfinamide on inhibition of Nav1.7 channel.Our study provides valuable insights into the structure–activity relationship of ralfinamide analogues towards Nav1.7 channel.It gives a strategy to improve the Nav1.7 inhibition of ralfinamide analogues.The compound 19h was more efficacious in antinociception in the mouse model of neuropathic pain induced by spared nerve injury compared with ralfinamide,although 19h shows a slightly shorter half-life and faster clearance liver microsomes.The open field test result suggests that the compound 19h had no alteration on locomotion in mice.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Financial supports by National Natural Science Foundation of China (Nos.82003565 and 81973162) and the Science and Technology Commission of Shanghai Municipality (No.20S11902300,China).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.08.026.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis