
The Pd-catalyzed synthesis of difluoroethyl and difluorovinyl compounds with a chlorodifluoroethyl iodonium salt (CDFI)
2022-06-18 10:53YaruNiuChengyaoKimmyCaoChenxinGeHongmeiQuChaoChen
Chinese Chemical Letters 2022年3期
Yaru Niu,Chengyao Kimmy Cao,Chenxin Ge,Hongmei Qu,Chao Chen,c,∗
a Key Laboratory of Systems Bioengineering,Ministry of Education,Department of Pharmaceutical Engineering,School of Chemical Engineering and Technology,Tianjin University,Tianjin 300350,China
b Key Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education,MOE),Department of Chemistry,Tsinghua University,Beijing 100084,China
c State Key Laboratory of Elemento-Organic Chemistry,Nankai University,Tianjin 300071,China
Keywords:Iodonium salts C-H activation Pd-catalyzed Difluoroethylation Difluorovinylation
ABSTRACT Herein,we report a simple and efficient method for the direct installation of chlorodifluoroethyl group onto aromatic molecules of various aromatic amides with a new 2-chloro,2,2-difluoroethyl(mesityl)iodonium salt (CDFI).Moreover,the chlorodifluoroethyl compounds could be smoothly converted into difluorovinyl compounds in a one-pot or discrete procedure and regarded as a steady source of difluorovinyl compounds with “HCl-mask”.
Due to the high electronegativity and small size of fluorine,fluorine and fluorine-containing substituents often provide advantageous properties to the molecules,such as modification of the electronic properties,improvement of the metabolic stability,and enhancement of the lypophilicity [1,2].By estimation,approximately 30% of all agrochemicals and 20% of all pharmaceuticals contain fluoro atom(s) [3,4].Among various fluorinated compounds,difluoroethylated (-CH2CF2-) and difluorovinylated (-CH=CF2) groups are both crucial pharmaceutical moieties and precursors of drugs,and the difluorovinylated moiety is regarded as an important carbonyl bioisostere (Fig.1) [5–11].Hence,the development of a convenient and efficient reaction to introduce difluoroethyl or difluorovinyl groups has been attractive from the chemistry community.The difluoroethyl moieties are usually built from difluorination of carbonyl group with active fluodides (e.g.DAST-diethylaminosulfur trifluoride) or reduction of difluoro- actetate derivatives with boranes[12–18],which are not so efficient and straightforward.There some difluorovinyl reactions emerged,especially aided by transitionmetal catalysis besides the traditional Wittig type reaction of carbonyl compounds with Ph3P=CF2ylid (Scheme 1) [19–24].But still now,the development of effective introduction of difluoroethylated (-CH2CF2-) and difluorovinylated (-CH=CF2) moieties in organic compounds is highly desirable.Herein,we would like to present a new method to synthesize difluoroethyl compounds with a novel iodonium salt.Moreover,the newly formed difluoroethyl compounds could be directly eliminated to deliverd difluorovinylated (-CH=CF2) compounds (Scheme 1)in situor in a discrete procedure.
Nowadays,hypervalent iodonium reagents have experienced a rapid development and received considerable attention as mild,non-toxic and selective reagents in organic synthesis [25–27].And recently they have been effectively used in C-H functionalization reactions to introduce F-containing moieties,including CF3[28–31]or CH2CF3[32–35].During the research of the organic synthesis with hypervalent iodonium reagents in our group [36–42],we designed a new type of iodonium salt 1,which is apparently a useful source of -CH2CF2- group and provides a possibility for the further functionalization of the fluorinated products (Scheme 2).
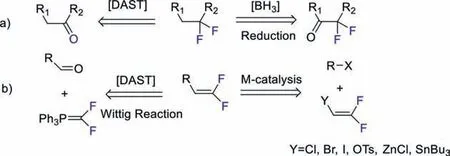
Scheme 1.Traditional synthetic routes to gem-difuorinated compounds.
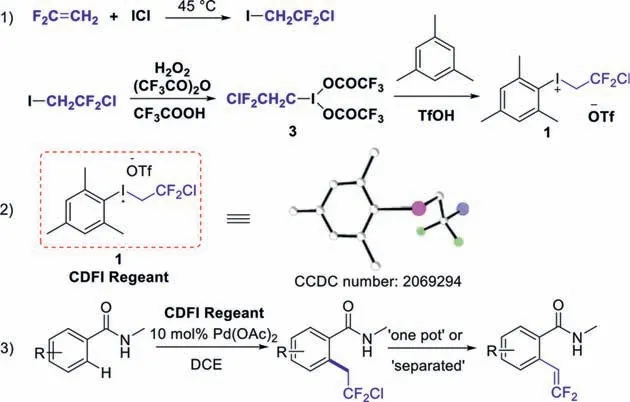
Scheme 2.The synthesis and structure of chlorodifluoroethyl iodonium salts and the application in the synthesis of difluoroethyl and difluorovinyl compounds.
The iodonium salt 1 was readily synthesized from 1-chloro-1,1-difluoro-2-iodoethane (ICH2CF2Cl),which could be prepared from iodine chloride (ICl) with 1,1-difluoroethene (CH2=CF2)[43,44].Subsequently,ICH2CF2Cl was oxidized by trifluoroperacetic acid preparedin situto gained the key intermediate (2-chloro-2,2-difluoroethyl)-λ3-iodanediyl bistrifluoroacetate 3.Then,the intermediate 3 was changed the ligand with mesitylene in the presence of triflic acid (TfOH) to generate 2-chloro-2,2-difluoroethyl(mesityl)iodonium triflate ([MesICH2CF2Cl]+[OTf]−) 1 in excellent yield.
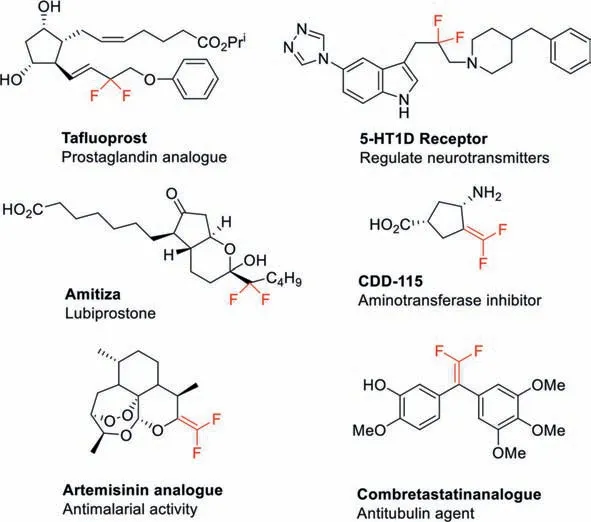
Fig.1.Some pharmaceutical molecules containing difluoroethylated (-CH2CF2-) and difluorovinylated (-CH=CF2) moieties.
As is shown in Scheme 2,the structure of the iodonium salt is established by X-ray crystallography.The single crystal could be obtained by slow evaporation of its diethyl ether- dichloromethane solution.The crystal data shows that the C(CH2)-I bond is 2.317 ˚A and the C(CF2)-Cl bond 1.778 ˚A.Due to the presence of the sterically hindered mesityl group,the bond length of the other C(Ar)-I bond is 2.129 ˚A,which seems more difficult to react.Therefore,this reagent could be used as a general -CH2CF2- source,which had the possibility for the further functionalization.
With the iodonium salt 1 in hand,we turn to investigate the chlorodifluoroethylation of 4-(tert-butyl)-N-methylbenzamide 2a as the model substrate in the presence of palladium catalyst.To testthe reaction feasibility,we screened different solvents,palladium catalysts and acid additives to find the optimal reaction conditions for this transformation (Table 1).The reaction of 2a with the iodonium salts in dry dichloroethane (DCE) without catalyst gave no product (entry 1).Hence,various palladium catalysts,such as PdCl2,Pd2(dba)3,Pd(TFA)2and Pd(OAc)2were examined.As is shown in Table 1,the result with Pd(OAc)2as catalyst provided theo-substituted product 4a in 93% yield (entries 2–5).However,in the presence of palladium acetate,the chlorodifluoroethylation reaction could not occur without acid additives (entry 6).This indicated that the choice of acid additives was also critical,and then the acetic acid (AcOH) as an additive was tested,which afforded only trace product 4a (entry 7).Whenp-toluenesulfonic acid monohydrate (TsOH·H2O) and boron trifluoride ethyl ether(BF3·Et2O) were employed,the product can be delivered in a moderate yield (entries 8 and 9).Pleasingly,trifluoroacetic acid (TFA)was found to be an ideal additive to afford the expected product in 93% yield (entry 5).Other solvents,such as methanol (MeOH),tetrahydrofuran (THF),toluene (Tol),dichloromethane (DCM),and dichloroethane (DCE) were also screened for optimized conditions,and it was observed that DCE afforded better yield than the other solvents (entries 10–13).After extensive evaluation,we established that a combination of Pd(OAc)2(10 mol%),trifluoroacetic acid (TFA)(1.2 equiv.),and iodonium salt (1.2 equiv.) in DCE at ambient temperature was optimal and produced the desired chlorodifluoroethylation product (4a) in 93% yield.
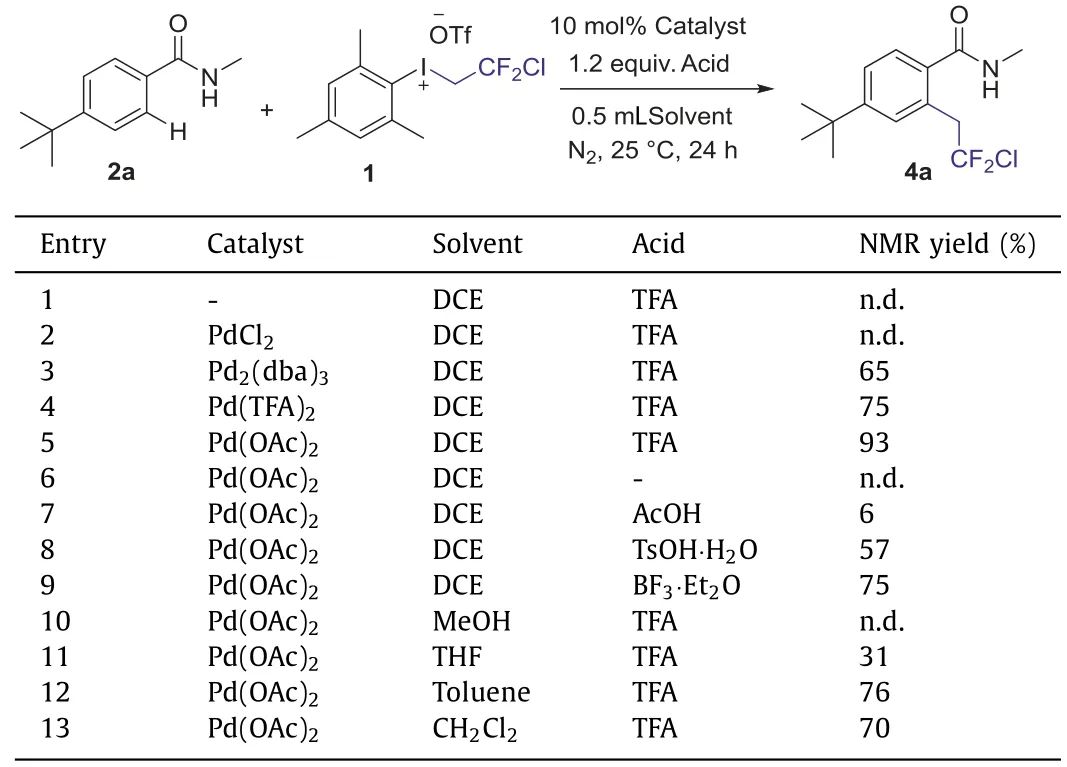
Table 1 Optimization of the reaction conditions.
With the optimal conditions in hand,the generality of this transformation was then examined with different aryl-substituted benzamides (Scheme 3).It was found that electron-donating methyl and methoxy groups ino-,m-andp-positions were well tolerated and the corresponding products formed in good to excellent yields (4b-4f).Moreover,benzamides with either atertbutyl or phenyl group all worked well to afford products 4a or 4t in 87%,75% yields,respectively.Chloromethyl substituent at thep-position was also tolerable,rendering the corresponding products 4g in 66% yield.Remarkably,theN-methylbenzamides bearing halogen substitutions such as fluoro (4h,4o),chloro (4i,4l,4p),bromo (4j,4m) and iodo (4k) groups could be well introduced to the 4-,5-,6-positions of the aryl ring in the present system,and all afforded the target products in satisfied yields.More appealingly,electron-withdrawing groups including ester,trifluoromethyl,and trifluoromethoxy groups were remarkably suitable for the reaction,which led to theo-chlorodifluoroethylated products in moderate to good yields (4n,4q,4r).Further,multisubstitutedN-methylbenzamide also reacted well and resulted in 80% yield (4s).Notably,naphthamide were also effectively converted into their corresponding products,and the yields of 4u and 4v were 57% and 85%,respectively.Moreover,chlorodifluoroethylation of heterocyclic thiophene structure (4w,4x),including no substituent and a chloro-substituted thiophene,were also converted smoothly into the desired products.Finally,the structure of 4u was confirmed by X-ray diffraction analysis (CCDC: 2064869,see Supporting information for more details).
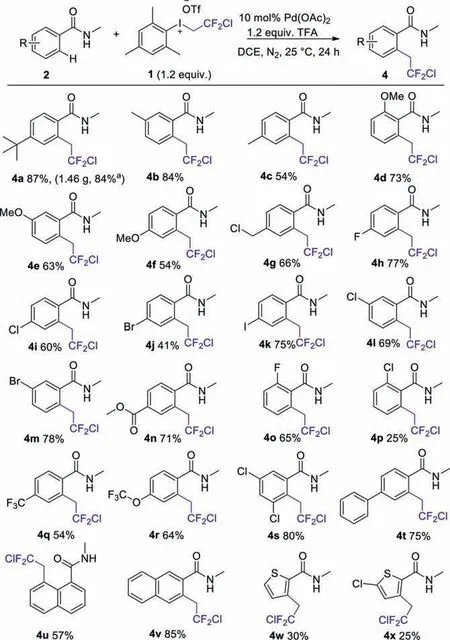
Scheme 3.The scope of chlorodifluoroethylation reaction of various benzamides 2 with CDFI 1.Reactions were performed on a 1.0 mmol scale;isolated yield.a Reactions were performed on a 6 mmol scale.
Next,we investigated a variety of diverseN-substituted handles under optimal conditions to further broaden the substrate scope(Scheme 4).First,we explored the application ofN-substituted amide equipped with isopropyl,tert-butyl,phenethyl and cyclohexyl substituents on the nitrogen atom.To our delight,electrondonating and electron-withdrawing groups on the aryl ring were well-tolerated in moderate to good yields (6a-6e,62%–86%).Notably,N,N-disubstituted substrates including 6f and 6g also reacted well in 43% and 66% yield,respectively.Moreover,quinazolinone derivatives with 2-position substituted with phenyl group could also be applied in this system to gave the Pd-N coordinated cyclopalladium complex catalyzed product 6h and the structure was confirmed by the X-ray diffraction analysis (CCDC: 2094743,see Supporting information).

Scheme 4.The scope of chlorodifluoroethylation reaction of various benzamides 5 with CDFI 1.Reactions were performed on a 1.0 mmol scale;isolated yield.
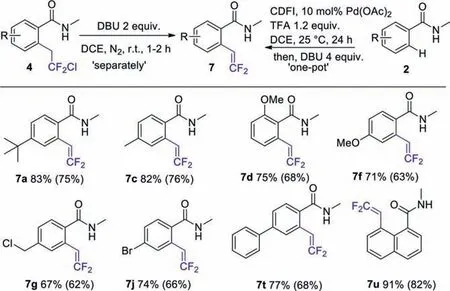
Scheme 5.The scope of elimination reaction of various benzamides 4 and 2.Reactions were performed on a 1.0 mmol scale.Isolated yield (in bracket were that performed on a 0.5 mmol scale).
The broad functional group tolerance of this palladium catalyzed chlorodifluoroethylation reaction gave facilely access to difluorovinyl derivativesviaelimination reaction (Scheme 5).When 2 equiv.of DBU was added to the DCE solution of the chlorodifluoroethylated substrates,the dehydrochlorination products 7 were formed in high yields at room temperature.Most of the substrates were able to undergo dehydrochlorination to provide the corresponding products in outstanding yields,such as methyl (7c,82%),chloromethyl (7g,67%),methoxy (7d,7f,75%,71%),tert-butyl(7a,83%) and phenyl (7t,77%).In particular,product 7u could be readily dehydrochlorinated to give 8-(2,2-difluorovinyl)-N-methyl-1-naphthamide in 91% yield.It is worthy to note that product 7j was obtained in 74% yield and the molecular structure of 7j was unambiguously established by X-ray crystallography (CCDC:2064872,see Supporting information for details).
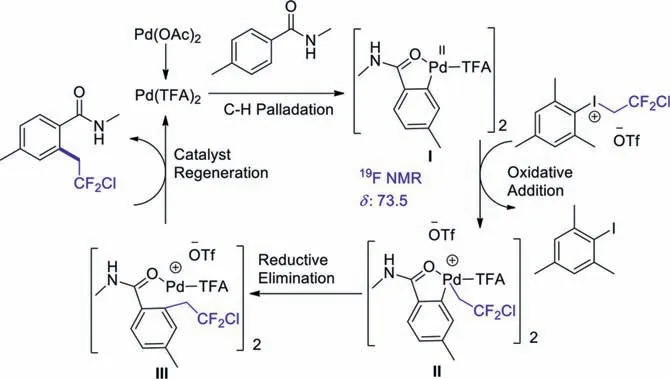
Scheme 6.A plausible mechanism.
This procedure enables us to regard the chlorodifluoroethylated products as “HCl-masked” difluorovinyl derivatives,since most of the difluorovinyl compounds are relatively reactive and can not be stored for long time due to its high activity to undergo polymerization [45–47].In addition,the difluorovinyl compounds could be made in a one-pot procedure.Thus,DBU (4 equiv.) was added to the reaction mixture of benzamides with chlorodifluoroethyl iodonium salts and sequentially the difluorovinyl compounds were isolated in comparable yields.
Having established the substrate generality of the reaction,we conducted related experiments to understand the mechanism of this transformation.At first,we tried to monitor the cyclopalladated species by19F NMR under the reaction conditions,which is a critical intermediate in the reaction [48,49].The formation of the intermediate I was isolated and characterized by19F NMR spectroscopy (δ−73.5).Then we detected the stoichiometric reaction of the cyclopalladated species I with the treatment of 2-chloro-2,2-difluoro-ethyl(mesityl)iodonium salt.To our delight,the expected chlorodifluoroethylated products were obtained rapidly at room temperature.Based on these results,we proposed the following mechanism (Scheme 6).First,in the presence of trifluoroacetic acid (TFA),the reaction was initiated through the palladium acetate activation process.Then,the palladium species were able to construct a cyclometalated intermediate I with the guidance of directing group.Subsequently,the complex I was chlorodifluoroethylated by CDFI 1 to form the intermediate II.Finally,the CH2CF2Cl on the Pd(IV) center was transferred to the aromatic ring to produce intermediate III,which experienced the elimination reaction to afford the product and regenerated the active Pd(II)species for the next catalytic cycle.
In conclusion,we have developed a novel CDFI reagent 1 and used it for chlorodifluoroethylation of C−H bond inNmethylbenzamides by the Pd-catalyst.Remarkably,the highly efficient iodonium salt,the broad scope of amides,and the mild reaction conditions make our methodology extraordinarily significant in drug discovery and organic synthesis.Furthermore,the value of this methodology is further demonstrated by the facile removal the HCl mask and efficient transformation to difluorovinyl products.And the further application of this 2-chloro,2,2-difluoroethyl(mesityl)iodonium salt is currently ongoing in our laboratory.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (No.2016YFB0401400),the National Natural Science Foundation of China (Nos.22071129,21871158 and 21672120).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.09.004.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis