
Theoretical and experimental investigations of the enantioselective epoxidation of olefins catalyzed by manganese complexes
2022-06-18 10:53JinLinFngWngJingTinJishengZhngYongWngWeiSun
Chinese Chemical Letters 2022年3期
Jin Lin,Fng Wng,Jing Tin,Jisheng Zhng,Yong Wng,Wei Sun,∗∗
a State Key Laboratory for Oxo Synthesis and Selective Oxidation,Center for Excellence in Molecular Synthesis,Suzhou Research Institute of LICP,Lanzhou Institute of Chemical Physics (LICP),Chinese Academy of Sciences,Lanzhou 730000,China
b Institute of Drug Discovery Technology,Ningbo University,Ningbo 315211,China
c University of Chinese Academy of Sciences,Beijing 100049,China
Keywords:Epoxidation of olefins Manganese complexes Metal oxo Enantioselective DFT
ABSTRACT The enantioselective epoxidation of olefin by MnII(R,R-PMCP)(OTf)2,H2O2 and H2SO4 was explored by DFT calculations and experiments.Theoretical results suggest that [MnV(O)(R,R-PMCP)(SO4)]+ species with a triplet ground spin state serves as the active species for the olefin epoxidation.It can be generated by the H2SO4 assisted O-O heterolysis of MnIII(OOH) species.MnIII-persulfate is also involved in this system,but it cannot promote the olefin epoxidation directly,preferring instead to transform into MnV(O).Actually,the asymmetric epoxidation reactions with H2O2/H2SO4 or Oxone provide similar enantioselectivity in the presence of manganese catalyst.These observations further support the transformation of MnIII-persulfate to MnV(O) species.
Developing sustainable and efficient catalysts for the oxidation of hydrocarbons under mild conditions has attracted considerable interests in synthetic chemistry and the chemical industry [1–5].In this regard,metalloenzymes utilize earth-abundant metal center(e.g.,iron or manganese) to catalyze a wide range of oxidation reactions by activating dioxygen under mild conditions,thereby providing an unique platform for the development of efficient oxidation catalysts [6,7].Over the past decades,a plethora of biomimetic catalysts capable of chemo- and enantioselectively oxidizing C=C and C-H bonds have been established [8,9].In particular,iron and manganese complexes with linear tetradentate nitrogen (N4) ligands inspired by the nonheme enzymes have been becoming the catalyst choice of chemo- and enantioselective oxidations,owing to their facile synthesis and diversification [10–12].In these catalytic systems,aqueous hydrogen peroxide (H2O2) usually serves as the terminal oxidant,thus making these systems green and sustainable property.It should be noted that carboxylic acid,such as acetic acid,is a pivotal additive for efficient oxidation catalysis with these iron and manganese complexes.
In the case of nonheme iron(II) complexes,the effect of acetic acid (AcOH) was originally observed by Jacobsen and coworkers in the study of practical epoxidation olefin with MEP-FeII(MeCN)2(SbF6)2/H2O2[MEP=N,N’-dimethyl-N,N’-bis(2-pyridylmethyl)-ethylene-1,2-diamine].Upon addition of catalytic amount of AcOH,FeII(MEP) complex displayed excellent reactivity in the olefin epoxidation [13].Later,Que and co-workers proposed a carboxylic acid assisted O-O bond cleavage mechanism,in which a highly reactive FeV-oxo species was generatedviaa heterolytic O-O bond cleavage of an FeIII-OOH precursor in the presence of acetic acid [14].The carboxylic acid assisted mechanism in the catalytic epoxidation reactions promoted by nonheme iron catalyst/H2O2/carboxylic acid has been supported experimentally and theoretically [15,16].In particular,on the basis of density functional theory (DFT) calculations for FeII(S,S-PDP)/H2O2/AcOH catalytic system {PDP=2-[[2-(1-(pyridin-2-ylmethyl)-pyrrolidin-2-yl)pyrrolidin-1-yl]methyl]pyridine},the DFT investigations support the involvement of (S,S-PDP)FeIV(O)(AcO•) as oxidizing speciesviathe O-O bond homolysis from (S,S-PDP)FeIII(κ2-peracetate).This[oxoiron(IV)-AcO]cation radical has similar electronic structure as that of compound I of P450 [15].Actually,the multiple-oxidant nature has been observed in nonheme iron systems,largely depending on the binding ligand around iron center.Recently,an [FeV(O)(OAc)(PyNMe3)]2+species has been spectroscopically trapped by Costas and co-workers,which is capable of hydroxylating unactivated alkyl C-H bonds [17].Interestingly,Nam and co-worker have demonstrated that nonheme high-spin iron(III)-acylperoxo complex with 13-TMC ligand (13-TMC=1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclotri-decane) is strong oxidant capable of oxygenating hydrocarbons prior to their conversion into iron-oxo speciesviaO-O bond cleavage [18].Based on these achievements,DFT calculations have definitely helped to predict the possible active species and to address mechanistic issues,thereby elucidating the multiple-oxidant nature in the catalytic cycle of oxidation reactions in combination with the experimental studies.
In parallel to iron systems,nonheme manganese complexes bearing chiral N4 ligands have been shown to be effective catalysts in the asymmetric oxidations [10–12].In these manganese catalytic systems,carboxylic acids (e.g.,AcOH) still play a key role in the activation of H2O2together with the manganese complexes.In the past decade,Costas [19,20],Bryliakov [16,21]and our group[22–26]have developed a variety of nonheme manganese complexes for efficient oxidation catalysis.Most of this class of effective iron and manganese catalysts thus far possesses same feature that is supported by chiral N4 ligands,thus providing two labile cis-binding sites for H2O2activation.In these reactions,it has been proposed with indirect experimental evidence that the active intermediate is MnV-oxo species [16,27].Compared to well documented mechanism of nonheme iron catalysts,however,the mechanistic aspects concerning the manganese catalysts are underdeveloped.In this context,what kinds of manganese species will involve in the manganese/H2O2catalytic systems,MnV(O),MnIV(O) cation radical or other possible species [28]? An in-depth understanding of the mechanism is a fundamental task for rational ligand design toward tuning of enantio-control and reactivity.Previously,a variety of manganese complexes with aryl-substituted MCP (MCP=N,N’-dimethyl-N,N’-bis-(2-pyridinylmethylcyclo-hexane-1,2-diamine) N4 ligands by our group have been showed excellent enantiocontrol in the asymmetric epoxidation of olefins,in which sulfuric acid could significantly enhance the activity for olefin epoxidation [22,29,30].Continuing on this line of research,we report herein the systematic theoretical calculations on the epoxidation mechanism catalyzed by the manganese modeling catalyst,MnII(R,R-PMCP)(OTf)2with H2O2in the presence of sulfuric acid.Based on the DFT results,[MnV(O)(R,R-PMCP)(SO4)]+with a triplet ground spin state serves as the active species for the olefin epoxidation.Interestingly,a MnIII-persulfate is likely involved in this catalytic system,however,DFT results suggest that the energy barrier of its C=C double bond epoxidation (18.7 kcal/mol) is higher than that of its transformation to a [MnV(O)(R,R-PMCP)(SO4)]+species (9.5 kcal/mol).In addition,the direct use of oxone (potassium peroxymonosulfate,2KHSO5·KHSO4·K2SO4) as oxidant also provides a comparable enantioselectivity as that of H2O2/sulfuric acid system.
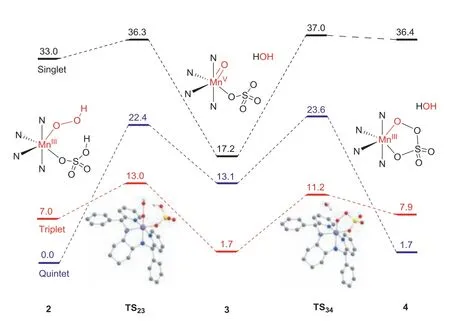
Fig.1.Potential energy profiles for the conversion of MnIII-OOH to manganic persulfate via MnV-oxo at B3LYP/Def2-TZVPP//TZVP(Mn,OOH,HSO4,N atoms in the ligand)-6-31G∗(C and H atoms in the ligand) level including zero-point energy and solvation corrections.
High-valent metal-oxo species are highly reactive intermediates involved in the oxidation of organic substrates by heme and nonheme enzymes and their model compounds.In the biomimetic manganese catalyst/H2O2/acid catalytic system,MnV-oxo,MnIV-oxo and MnIII-peroxo may be involved as the possible active species as those of nonheme iron catalytic systems.As previously proposed by Bryliakov/Talsi and co-workers,still,the manganese catalytic system started from an MnIII-hydroperoxide species,followed protonation of the hydroperoxide ligand by the coordinated acid in MnIII-hydroperoxo intermediates facilitates the O-O bond cleavage,generating MnV-oxo species as reactive epoxidizing intermediates.As shown in Scheme 1,possibly,[MnV(O)(R,R-PMCP)(SO4)]+3,MnIII-persulfate 4 can be generated from MnII(R,R-PMCP)(OTf)21 with H2O2as the oxidant in the presence of sulfuric acid.Previously,we have briefly studied the formation of[MnV(O)(R,R-PMCP)(SO4)]+3 from its precursor,MnIII-OOH species(2) by DFT calculations [29].The results showed that the transformation occurs reasonablyviaan O-O bond heterolysis by the aid of coordinated sulphuric acid.For this process,as shown in Fig.1,the calculated ground state for complex 2 is the quintet spin state.In addition,low-spin singlet 2 is involved,however,the singlet state is more than 33.0 kcal/mol higher in energy than quintet one.In the first step,cleavage of the O-OH bond of 2 leads to the formation of Mn-oxo 3,in which a spin change from the higher quintet spin to triplet spin state occurs (two-state reactivity) [31,32].In this case,the transition state(TS) TS23 in triplet and quintet states are found to be 13.0 and 22.4 kcal/mol,respectively.Possibly,cyclic MnIII-persulfate is involved as previously reported (S,S-PDP)FeIII(κ2-peracetate) species.Actually,DFT calculations support the formation the MnIIIpersulfate through the O-O bond coupling reaction with a barrier of 11.2 kcal/mol.The potential energies of triplet MnV-oxo (33) and quintet MnIIIpersalt (54) are equal,indicating that both species can possibly exist in the reaction system and act as epoxidizing reagent.
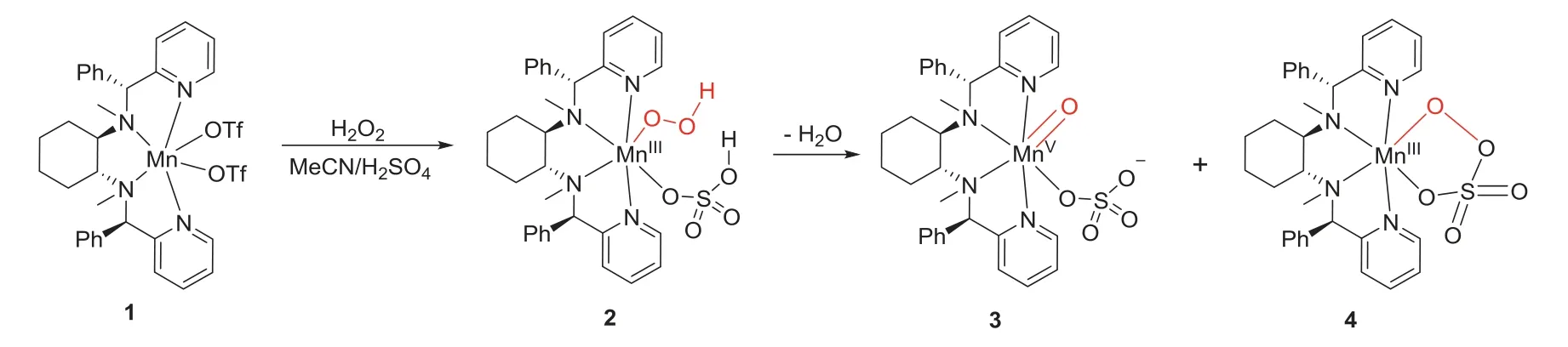
Scheme 1.Possible intermediates for the MnII(R,R-PMCP)(OTf)2 with H2O2 in the presence of sulfuric acid.
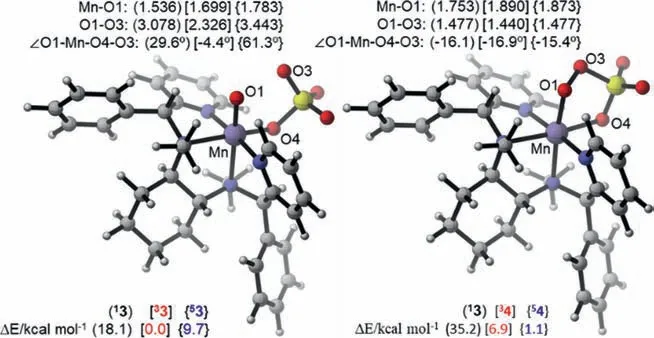
Fig.2.Key geometric parameters and spin-state energies of MnV(O) 3,MnIIIpersulfate 4.
Fig.2 shows the key geometric parameters and spin-state energies of MnV(O) 3,MnIII-persulfate 4 without the resulting H2O.The bond length of Mn–O in33 is 1.699 ˚A,similar to the Mn-O bond length (i.e.,1.71 ˚A) of an S=1 MnVO complex,[(S-PMB)MnV(O)(OAc)]+(S-PMB=(S)-N-methyl-1-(1-methyl-1Hbenzo-[d]imidazol-2-yl)-N-((1-((1-methyl-1H-benzo[d]-imidazol-2-yl)methyl)pyrroli-din-2-yl)-methyl)methanamine) [33].Especially,the33 has the shortest distance of O1-O3,providing a justification of the lowest transition state in the formation of MnIII-persulfate 4 than those of singlet- and quintet states (2.326,3.078 and 3.443 ˚A).
Then,asymmetric epoxidation by Mn-oxo 3 was calculated,styrene was chosen as a model substrate.As shown in Fig.3,styrene can bind to the Mn-oxo 3 and two diastereomeric complexes form with different stabilities.In the case of the triplet 3,the energy difference in both diastereomeric complexes is 2.0 kcal/mol.In the first step,a radical intermediate (IM3) is generated after the reaction of Mn-oxo 3 with styrene.The profile shows that this step has the lowest energy barrier of 2.6 kcal/mol on the triplet surface.The calculated results suggest this step is the enantioselectivity-determining step.The relative free energy of TS1(R) is 0.5 kcal/mol higher than that of TS1(S),which suggests the major epoxide product isS-configuration.This result is also agreement with the experimental observation.Inspection of the geometric details of preferred TS1 demonstrates that the phenyl group on the PMCP ligand plays a key role in tuning the enantiocontrol of the reaction.Bulkier group will lead to higher enantioselectivity,for example,(R,R)-DBP-MCP-Mn(OTf)2having 3,5-ditert-butyl-phenyl groups on the ligand provide a better ee value(Table 1,60%ee vs.50%ee) [29].When (R,R)-MCP-Mn(OTf)2complex,without phenyl group,was used as catalyst in the epoxidation reaction,it provided a poor yield.Consistently,the formationof MnV-oxo from (R,R)-MCP-Mn(OTf)2has an increased energy barrier of 15.1 kcal/mol (Fig.S1 in Supporting information),compared to that of (R,R)-PMCP-Mn(OTf)2(13.0 kcal/mol).Finally,the IM3 undergoes an intramolecular ring closure to produce the epoxide.This step also including a spin state change from triplet to quintet.Besides,to gain more insight into the possible MnIII-persulfate 4,we have computationally examined the epoxidation styrene by 4 (Fig.4).The styrene epoxidation by 3 is exothermic in each step,while the epoxidation by 4 is endothermic in the first step.Besides,this pathway has an energy barrier of 18.7 kcal/mol,even much higher than that of transformation of 4 to Mn-oxo 3 (9.5 kcal/mol).Taken together,these results imply that MnIII-persulfate 4 is not an epoxidizing species.Instead,it may serve as a reservoir for the formation of reactive MnV(O) species during the oxidation reactions.In fact,the direct use of Oxone as an oxidant also provided a comparable enantioselectivity as that of H2O2in the presence of sulfuric acid,when (R,R)-DBP-MCP-Mn(OTf)2was used as the catalyst(Table 1,entries 5 and 6) [34,35].The lower yield attributes to its bad solvability in organic solvent,despite water is used as a solvent.
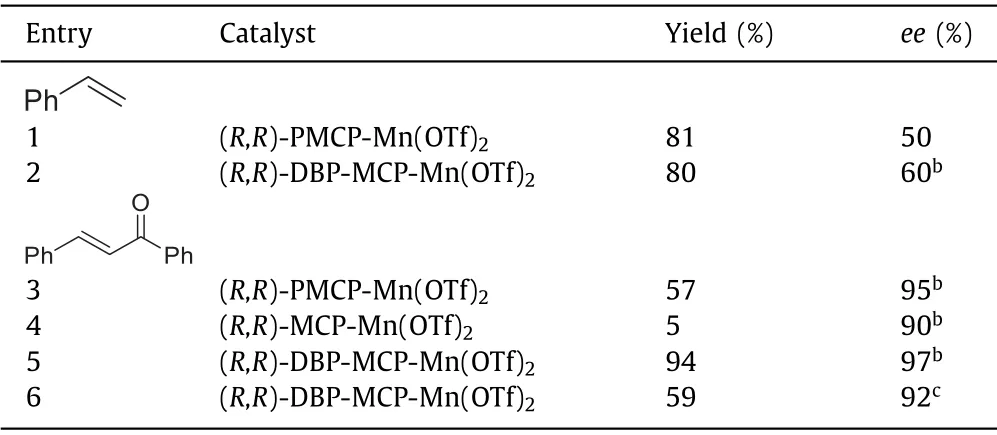
Table 1 Asymmetric epoxidation of olefins.a
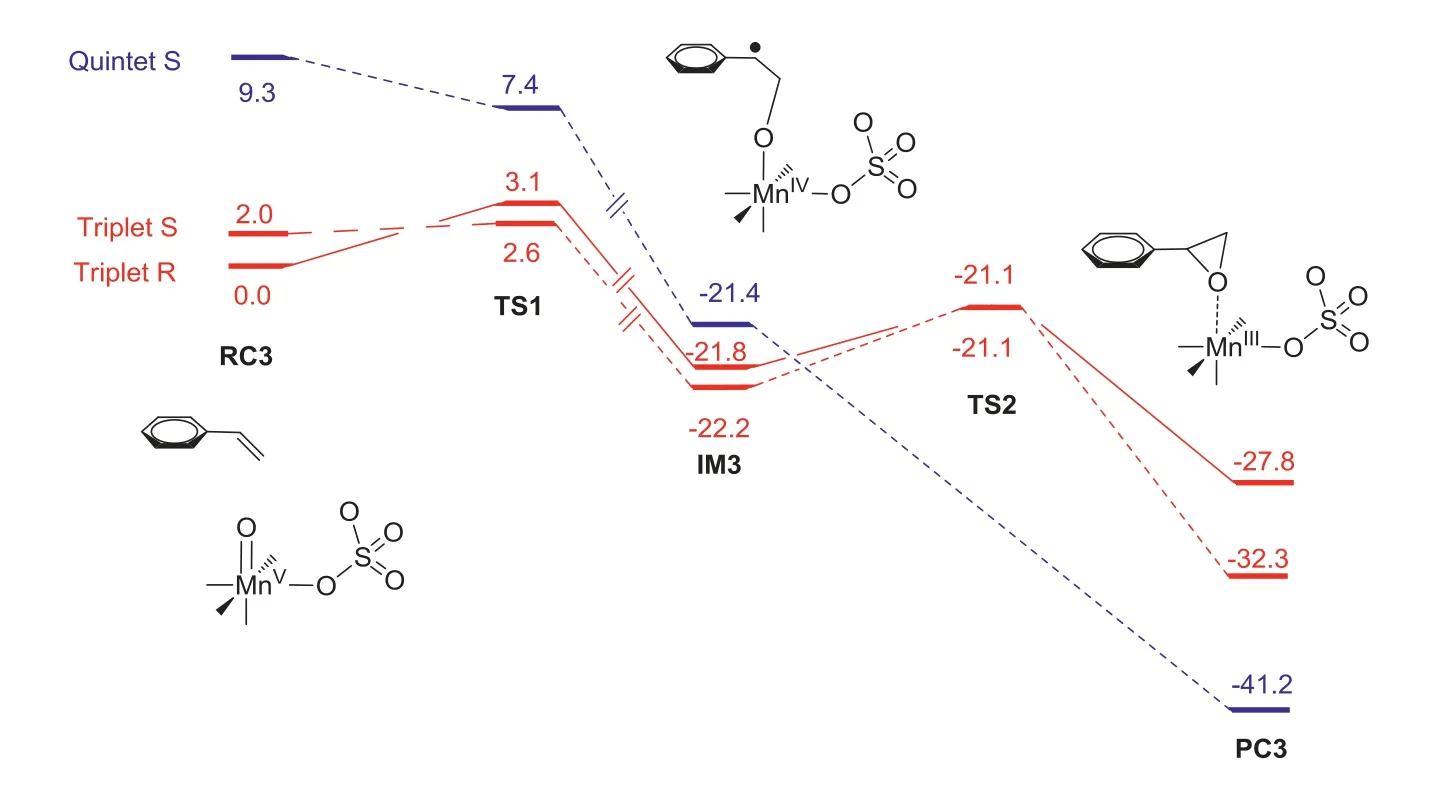
Fig.3.Energy profiles for the styrene epoxidation by MnV-oxo at B3LYP/Def2-TZVPP//TZVP(Mn)-6-31G∗(other atoms) level including zero-point energy and solvation corrections.
To further understand the enantioselectivity,the independent gradient model (IGM) [36]ofS-3TS1 andR-3TS1 was established to elucidate the possible interactions.As shown in Fig.5,theSisomer has an additional T-shapedπ-π(Tπ-π) between phenyl on the ligand and the styrene,while theRisomer has only paralleldisplacedπ-π(PDπ-π) interaction between the pyridine of the ligand and styrene [37].Besides,the deformation energy ofS-3TS1 isomer is about 1.6 kcal/mol lower than that ofRisomer.Taken together,the additional T-shapedπ-πinteraction and less deformation make theS-3TS1 more favored.
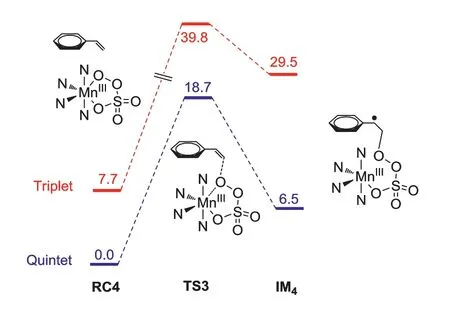
Fig.4.Energy profiles for the styrene epoxidation by MnIII-persulfate at B3LYP/Def2-TZVPP//TZVP(Mn)-6-31G∗(other atoms) level including zero-point energy and solvation corrections.

Fig.5.IGM for two isomers of triplet TS1.
In summary,DFT calculations were performed to investigate the mechanism of the asymmetric epoxidation of olefin by (R,R)-PMCPMn(OTf)2/H2O2/H2SO4catalyst system.The computational results suggest that [MnV(O)(R,R-PMCP)(SO4)]+is an epoxidizing reagent,and it promotes the epoxidation reaction with a low barrier.A cyclic MnIII-persulfate species is also involved in this catalyst system,however,it cannot participate in the epoxidation directly due to the higher energy barrier.
Declaration of competing interest
The authors declare that they have no conflicts of interest to this work.
Acknowledgment
We acknowledge financial support of this work from the National Natural Science Foundation of China (Nos.21773273,21802153).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.08.092.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis