
Nanoplasmonic zirconium nitride photocatalyst for direct overall water splitting
2022-06-18 10:52YuLiuXioweiZhngLishLuJunYeJinlinWngXiominLiXuedongBiWenlongWng
Chinese Chemical Letters 2022年3期
Yu Liu,Xiowei Zhng,Lish Lu,Jun Ye,Jinlin Wng,Xiomin Li,Xuedong Bi,c,Wenlong Wng,c,∗
a Beijing National Laboratory for Condensed Matter Physics and Institute of Physics,Chinese Academy of Sciences,Beijing 100190,China
b School of Physical Sciences,University of Chinese Academy of Sciences,Chinese Academy of Sciences,Beijing 100190,China
c Songshan Lake Materials Laboratory,Dongguan 523808,China
Keywords:Zirconium nitride Surface plasmon Noble–metal–free Photocatalytic water splitting Hot electron
ABSTRACT The ability of plasmonic nanostructures to efficiently harvest light energy and generate energetic hot carriers makes them promising materials for utilization in photocatalytic water spitting.Apart from the traditional Au and Ag based plasmonic photocatalysts,more recently the noble–metal–free alternative plasmonic materials have attracted ever–increasing interest.Here we report the first use of plasmonic zirconium nitride (ZrN) nanoparticles as a promising photocatalyst for water splitting.Highly crystalline ZrN nanoparticles with sizes dominating at 30–50 nm were synthesized that exhibit intense visible and near–infrared absorption due to localized surface plasmon resonance (LSPR).Without utilizing any noble metal cocatalysts such as Pt,the plasmonic ZrN nanoparticles alone showed stable photocatalytic activity for H2 evolution in aqueous solution with methanol as sacrificial electron donor.The addition of a cobalt oxide (CoOx) cocatalyst can facilitate the separation of photogenerated charge carriers and further improve the photocatalytic activity.The optimized CoOx modified ZrN photocatalyst was observed not only to activate the O2 evolution reaction with presence of electron acceptor,but also to drive overall water splitting for the simultaneous H2 and O2 evolution in the absence of any sacrificial agents.
The utilization of solar energy to drive photocatalytic water splitting for sustainable hydrogen production is one of the most prevailing ways for meeting the future need of environmentally friendly and renewable energy sources [1–8].With the use of noble metal Au and Ag plasmonic nanostructures,the field of plasmonic photocatalysis has witnessed tremendous progress over the past decade.As compared to the conventional semiconductor photocatalysts,the localized surface plasmon resonance (LSPR) in metal nanostructures provide a more viable way in high–efficiency harvesting of light energy,and especially the less energetic visible photons,for driving photocatalytic water splitting reactions[3,9].The plasmonic enhancement of photocatalytic water splitting involves complex mechanisms.In a typical metal–semiconductor heterojunction configuration such as the best studied Au–TiO2system,the enhancement mechanisms may include enhanced light absorption,forced separation of electron–hole pairs,plasmon–induced resonance energy transfer,and in particular,the transfer of plasmonic hot carriers from metal to semiconductor through a Schottky barrier [10].Meanwhile,as a simpler scenario,there are also studies that do not utilize a Schottky junction to drive the direct plasmonic photocatalysis including water splitting [11,12].
In recent years,the conductive transition–metal nitrides [13,14],such as TiN and ZrN,have undergone a resurgence of interest as promising alternatives to the conventional Au and Ag based noble metal plasmonic materials.TiN and ZrN have dielectric permittivities with a zero crossover wavelength in the visible region,very similar to Au,thereby exhibit plasmonic properties comparable to those of Au in the visible and near–infrared (NIR) spectrum [15,16].Coupled with their high temperature durability,chemical stability and low cost,TiN and ZrN can replace and even outperform the traditional noble metals in many plasmonic devices and applications [16–18].More recently,the experimental realizations of a variety of TiN–based plasmonic and nanophotonic devices have been reported,and there are also studies of utilizing the plasmonic TiN nanostructures for photocatalytic and photoelectrochemical watersplitting [19,20].However,as compared to TiN,to date the utilization of the excellent plasmonic properties of ZrN for practice applications has been far less explored.Herein,we present the first demonstration of the capability of ZrN nanostructures as a promising plasmonic photocatalyst for water splitting.
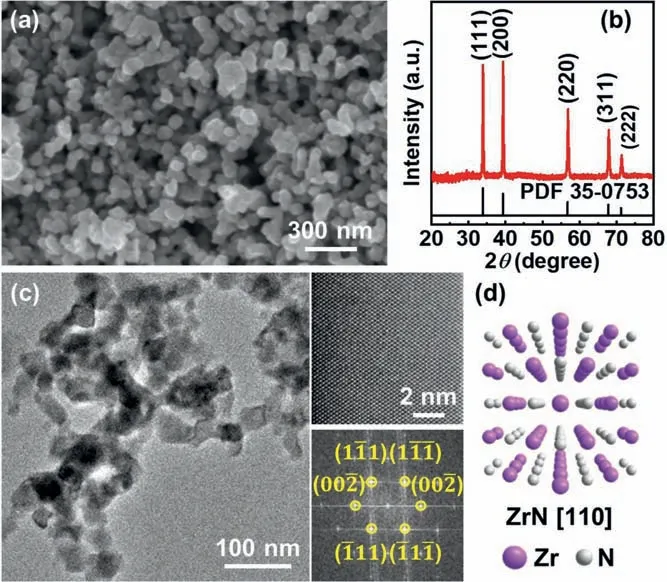
Fig.1.Morphology and crystal structure of zirconium nitride plasmonic nanoparticles.(a) SEM image of the as–synthesized ZrN nanoparticles with size distribution focusing at 30–50 nm.(b) XRD pattern of the ZrN powder.(c) TEM images of as–prepared ZrN nanoparticles.The insets are HRTEM image and the corresponding FFT pattern.(d) Atomic crystal structure of cubic ZrN viewed along the [110]direction.
As is known,the generation of hot carriers in Au nanostructures through nonradiative plasmon decay is driven by either intraband excitations within the conduction sp band or by interband excitations resulting from transitions between the filled d–band and sp band (Fig.S1) [21,22].In the cases of TiN and ZrN,however,the situation is a little more complicated.Due to the strong hybridization of Ti/Zr d orbitals with N p orbitals,the plasmonic optical properties involve the intraband excitations due to conduction electrons in the partially filled d band of Ti/Zr and the interband transition between N p to Ti/Zr d orbitals (Fig.S1 in Supporting information) [23,24].By comparison,the interband p→d transition energy of ZrN is larger than that of TiN.This means that ZrN will exhibit blue–shifted and stronger plasmonic response [14,25],thereby favoring the production of higher energy hot charge carriers.Keeping this mind,ZrN is expected to be a more favorable choice for better photocatalytic performance,and in particular,for achieving the overall water splitting,by which H2and O2are produced simultaneously over the photocatalyst.In our present study,this unique benefit of ZrN was proven.The plasmonic ZrN nanoparticles,with optimal modification of a cobalt oxide (CoOx) cocatalyst,exhibit photocatalytic activity for overall water splitting for the simultaneous H2and O2evolution.
Experimentally,ZrN nanoparticles in the form of free–standing powders are synthesized by controlled nitridation of ZrO2nanoparticles precursor with ammonia gas at elevated temperature (see Supporting information for experimental details).Shown in Fig.1a is a representative scanning electron microscope (SEM) image of the as–grown ZrN nanoparticles.Highly–uniform nanoparticles with irregularly rounded shape and size distribution focusing at 30–50 nm can be clearly visualized (particle size statistics are shown in Fig.S2 in Supporting information).Powder x–ray diffraction (XRD) (Fig.1b) demonstrated that the nanoparticles have the cubic rocksalt–type structure corresponding to a stoichiometric ZrN.It is important to note that,due to the extremely high nitridation reaction temperature (1100 °C),the problem of thermal sintering ZrN nanoparticles during growth can not be fully avoided.Nevertheless,the as–grown ZrN nanoparticles can be well–dispersed in aqueous solution and some other solvents with the aid of mild ultrasonication.Further transmission electron microscope (TEM) characterizations also revealed the structure uniformity of the crystalline ZrN nanoparticles,as shown in Fig.1c.The lattice–resolved high–resolution TEM (HRTEM) image in Fig.1c along with the corresponding fast–Fourier transform (FFT) pattern verified the single–crystallinity and phase purity of the cubic ZrN phase (Fig.1d).

Fig.2.(a) HAADF image of the as–prepared ZrN nanoparticles with a zirconium oxide shell.(b) HAADF image of a representative ZrN nanoparticle (white) and corresponding EDX mappings showing the distribution of nitrogen (blue),oxygen (red)and zirconium (green).(c) Experimental measured optical absorption spectrum of as–prepared ZrN nanoparticles in aqueous solution.The inset is the photograph of the corresponding aqueous dispersion.(d) FDTD Simulated absorption spectra of spherical 30–nm–diameter ZrN nanoparticle with a bare surface (red) and coated with a 3 nm ZrO2 shell (blue).
In previous studies of the growth of TiN and ZrN in the forms of thin films,coatings and nanoparticles,it has been well-established that a self–passivating native oxide layer with thickness of a few nanometers will be naturally formed at their when exposed to ambient atmosphere [26].The presence of this conformal protective surface shell is beneficial to the long-term durability of TiN and ZrN nanostructures at room temperature,while preserving their intrinsic plasmonic properties.Shown in Fig.2a is a high resolution high–angle angular dark field scanning transmission electron microscopy (HAADF–STEM) image of an individual ZrN nanoparticle in our present study,where a conformal surface shell with thickness ofca.3 nm can be clearly observed.Further element mapping by energy dispersive X–ray (EDX) spectroscopy (Fig.2b) reveals a distinct oxygen segregation at the nanoparticle surface,confirming that the surface layer is composed of zirconium oxide,presumably in the form of insulating ZrO2or substoichiometric ZrO2−x[27,28].According to the general consensus from previous literature [29],for plasmonic applications the existence of such an ultra-thin insulating surface layer can allow for the transfer of photogenerated charge carriers through electron tunneling.Fig.S3 (Supporting information) shows the X-ray photoelectron spectroscopy (XPS) measurement results of ZrN nanoparticles.The broad band of photoelectron emissions across Fermi level can be well discerned from the XPS valance band spectrum,verifying typical metal-like electronic structure of ZrN nanoparticles,albeit the presence of a significant amount of Zr-O bonding states at the nanoparticle surfaces.
Optical features of the ZrN nanoparticles were analyzed by UV–vis–NIR absorption spectroscopy.Fig.2c shows the optical absorption spectrum of the aqueous dispersion of the as–synthesized ZrN nanoparticles,and the inset is a photograph of the corresponding aqueous dispersion.The absorption spectrum shows a broad peak centered at ∼690 nm originating from the plasmon resonance of ZrN.Spectral measurements on several other specimens revealed typical variations in peak wavelength less than 5 nm.Finite–difference time–domain (FDTD) simulations of the absorption spectra were carried out to compare with the experimental results.Fig.2d displays the simulated spectra of a spherical 30–nm–diameter ZrN nanoparticle with a bare surface and coated with a 3 nm ZrO2shell.As consistent with previous studies [30],it is quite noticeable that the presence of the oxide layer on the surface of ZrN nanoparticles leads to a distinct red–shift of the LSPR peak due to the higher refractive index of ZrO2with respect to the solvent.As compared to the simulated spectra,the experimentally observed spectrum exhibits apparently broadening and red–shifting of the absorption band,which is mainly resulted from the relatively large size distribution of ZrN nanoparticles,and more importantly,from the aggregation induced interparticle coupling of the surface plasmon oscillations.Note that,as evidenced from the TEM image in Fig.1c,even in the aqueous dispersion,not all ZrN nanoparticles are monodisperse and isolated,and a broad red–shifted absorption band is known to be characteristic of aggregates of plasmonic nanostructures [31–33].
In a procedure similar to previously established method [34,35],we examined photocatalytic H2evolution over the plasmonic ZrN nanoparticles in aqueous solution with the presence of menthol(10 vol%) as a sacrificial electron donor.Without the help of any noble metal catalysts such as Pt,the bare ZrN nanoparticles achieved steady H2production under UV–visible light illumination.A typical time courses of H2evolution is shown in Fig.3a(black dots).Continuous H2evolution with no apparent degradation of the ZrN nanoparticles was clearly observed from the beginning of the reaction,with a rate of 0.48 mmol g−1h−1.No N2evolution was observed for the present catalyst even after the extended period of irradiation.The total evolution of H2after 24 h was 345.6 μmol,exceeding the molar amount of the starting ZrN catalyst.The stable photocatalytic activity observed in bare ZrN nanoparticles implies that there is an efficient separation of photogenerated electron-hole pairs,as also further confirmed by photoelectrochemical characterization.As seen in the time course of photocurrent generation on ZrN nanoparticle photocathode under potentiostatic condition (Fig.S4 in Supporting information),a fast response to the switching of the light ON–OFF signal can be clearly observed.Interestingly,with the addition of 1.0 wt% Pt as cocatalyst,only very limited enhancement of activity was observed,as shown in Fig.3a (blue dots),clearly suggesting a negligible effect of Pt on H2evolution over ZrN.Production of H2was also observed when other electron donors such as Na2S and triethanolamine (TEOA) were used instead of methanol.When Na2S was used as the sacrificial agent,much improved H2yields were observed,whereas in the case of TEOA,slightly decreased activity was observed in comparison with that of methanol (Fig.S5 in Supporting information).
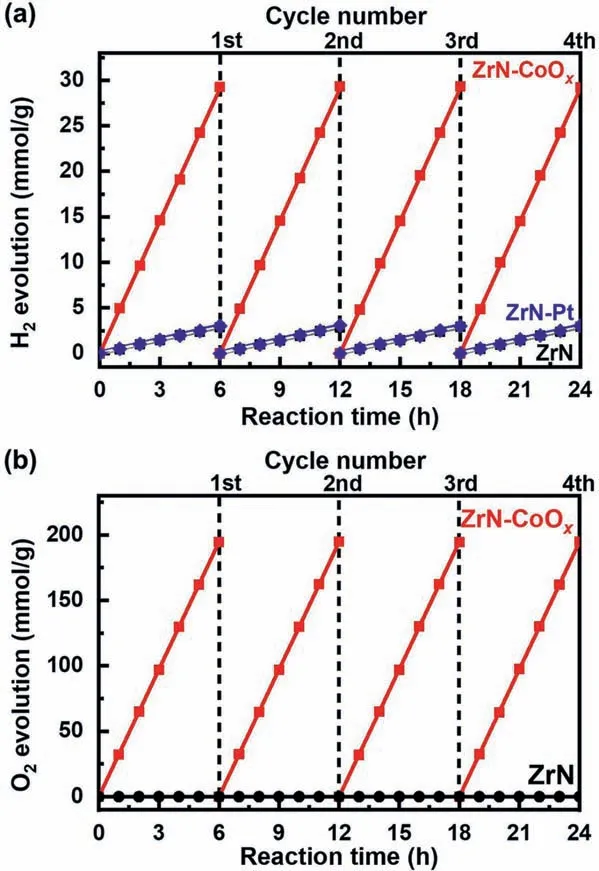
Fig.3.(a) Typical time courses of H2 evolution from water containing 10 vol%methanol as electron donor by ZrN (black dots,almost coincide with the blue dots),ZrN–Pt (blue dots) and ZrN–CoOx (red dots).(b) Typical time courses of O2 evolution from water containing 0.1 mol/L Na2S2O8 as electron scavenger by ZrN (black dots) and CoOx–modified ZrN (red dots).The reaction was continued for 24 h,with evacuation every 6 h (dashed line).
Of the two half–reactions involved in the splitting of water,water oxidation to produce molecular oxygen is mechanistically more challenging as it requires four positive holes and the formation of a new O–O bond.To overcome this rate limiting step,oxygen evolution cocatalysts (OEC) are commonly employed.Here in our present study,cobalt oxide (CoOx),which has been extensively used as OEC-cocatalyst for hole collectors in electrochemical and photocatalytic water splitting [36],was deposited on ZrN nanoparticles as OEC-cocatalyst to facilitate the separation of photogenerated charge carriers and further improve the photocatalytic activity.The deposition was achieved by an impregnation method from ethanol solution of Co(NO3)2,followed by NH3treatment at 700 °C and calcination at 200 °C in air [37].XPS measurements suggest that the CoOxcocatalyst is a mixture of Co2+and Co3+oxidation state (Fig.S6 in Supporting information).Whereas the bare ZrN catalyst was almost not active for water oxidation,producing only a trace of O2after 5 h of UV-visible illumination in an aqueous solution with Na2S2O8as electron scavenger,the oxygen evolution rate was dramatically increased when the CoOx–OEC was loaded.The optimal amount of CoOx–OEC in our present study is around 2.0 wt%,under which condition the O2–production rate can reach 32.44 mmol h−1g−1(Fig.3b).A further increase in the amount of CoOxaddition up to 3.0 wt% led to a slight decrease in activity,which may be caused by the agglomeration of CoOxinto larger nanoparticles resulting in less surface area.In addition,as compared to bare ZrN nanoparticles,a significant enhancement of H2evolution activity was also observed for CoOx–modified ZrN catalyst,with the H2evolution rate being ∼10 times higher in the aqueous methanol solution under the same UV-visible illumination(Fig.3a).

Fig.4.(a) Typical time courses of H2 and O2 evolution from water with 2.0 wt%by CoOx–modified ZrN catalyst in a pH 7 phosphate buffer.(b) Schematic of overall water splitting over the CoOx–modified plasmonic ZrN photocatalyst.
Given the enhanced photocatalytic performance of CoOx–modified ZrN photocatalyst,its photocatalytic performance for the overall water splitting was further examined.In a pH 7 phosphate buffer,the steady H2and O2evolution was observed on CoOx–modified ZrN without any sacrificial agents under UV-visible illumination.At lower CoOxaddition amount such as 0.5 wt%,the stoichiometric ratio of H2and O2was not equal to 2:1 and O2evolution rate is slower than the theoretical value (Fig.S7 in Supporting information).Nevertheless,an increase of the loading of CoOxcocatalyst to 2.0 wt% can give rise to more stoichiometric H2and O2production approximately equal to 2:1 (Fig.4a).For a direct comparison,the bare ZrN and Pt–modified ZrN were also tested but both of them showed no detectable H2or O2evolution in pure water.Fig.4b shows a schematic of the plasmonically driven water-splitting process occurring over the CoOx–modified ZrN photocatalyst.The optically excited surface plasmons in ZrN nanoparticles decay into hot electron-hole pairs.The CoOxcocatalyst acts as hole collector to prompt the separation of hot electron-hole pairs and also serves as active sites to activate the water oxidation reaction for O2evolution.At the same time,the spatially separated hot electrons are directly injected into water molecules to drive the H2evolution reaction.The net result of whole process is the direct plasmon-driven splitting of water.
We present the first demonstration of the use of plasmonic ZrN nanoparticles for direct hot electron–driven photocatalytic water splitting.The nanoplasmonic ZrN photocatalyst does not utilize any noble metal cocatalysts and can deliver stable photocatalytic activity for H2evolution in aqueous solution containing methanol as sacrificial electron donor.The modification of ZrN nanoparticles with CoOxcocatalyst can lead to significant enhancement of the H2evolution rate,and start up efficient O2evolution.More importantly,the direct overall water splitting for simultaneous H2and O2evolution can also be achieved by utilizing CoOxmodified ZrN photocatalysts in a pH 7 phosphate buffer without the requirement of any sacrificial agents.
Declaration of competing interest
The authors claim no conflicts of interest.
Acknowledgments
This work was supported by the Natural Science Foundation of China (Nos.21872172,21773303,51472267 and 51421002) and Chinese Academy of Sciences (Nos.XDB30000000,XDB07030100,Y8K5261B11 and ZDYZ2015-1).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.07.054.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis