
Bicyclic stapled peptides based on p53 as dual inhibitors for the interactions of p53 with MDM2 and MDMX
2022-06-18 10:52HongjinLiXiangyanChenMinghaoWuPanpanSongXiaZhao
Chinese Chemical Letters 2022年3期
Hongjin Li,Xiangyan Chen,Minghao Wu,Panpan Song,Xia Zhao,∗
a Key Laboratory of Marine Drugs,Ministry of Education,School of Medicine and Pharmacy,Ocean University of China,Qingdao 266003,China
b Laboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology,Qingdao 266237,China
Keywords:p53 MDM2 MDMX All-hydrocarbon stapling strategy Lactam stapling strategy Bicyclic stapling strategy
ABSTRACT In recent years,the strategy of inhibiting the interactions of p53 with murine double minute 2 (MDM2)and murine double minute X (MDMX) has been proved to be a promising approach for tumor therapy.However,the poor proteolytical stability and low intracellular delivery efficiency of peptide inhibitors limit their clinical application.Here,we designed and synthesized the bicyclic stapled peptides based on p53 by combining all-hydrocarbon stapling and lactam stapling strategies.We demonstrated that bicyclic stapled peptide p53-16 significantly improved α-helicity and proteolytic stability.Especially,p53-16 showed nanomolar binding affinity for MDM2 and MDMX.In addition,p53-16 could penetrate the cell membrane,and selectively inhibited the activity of tumor cells via activating p53 pathway in vitro.Our data suggest that p53-16 is a potential dual inhibitor of MDM2 and MDMX interactions.The bicyclic stapling strategy is a promising drug design strategy for protein–protein interactions inhibitors.
In recent decades,tumor has been one of the main causes of death in the developed countries,and has become a major public health problem worldwide [1].The tumor suppressor protein p53 is critical for maintaining genetic stability and preventing tumor development [2,3],and almost half of all tumors retain wild type(WT) p53.The most common impairment of p53 pathway is due to overexpression of its negative regulators,particularly murine double minute 2 (MDM2) and murine double minute X (MDMX) [4].MDM2,as a transcriptional target of p53 and E3 ubiquitin ligase,leads to the degradation of p53 proteasome and directly binds to the N-terminal of p53 transactivation domain (TAD) to regulate the levels of cellular p53 protein [5,6].MDMX,a homolog of MDM2,mainly inhibits the activity of p53 by binding with TAD and stimulates MDM2-mediated degradation of p53 [7].Thus,the inhibition of MDM2 and MDMX to reactivate WT p53 function is considered as a promising strategy for developing anticancer therapeutics [8].
According to the recent studies,dual inhibition of MDM2 and MDMX in tumor cells expressing WT p53 can activate p53 more effectively than antagonists that only inhibit MDM2 activity [9].So far,several small molecule dual inhibitors of p53–MDM2/MDMX interactions have been reported,such as an indolylhydantoin (RO-5963) [10];acis-imidazoline derivative (H109) [9];a tryptophanol-derived oxazolopiperidone lactam (OXAZ-1) [11];and a 1,4,5-trisubstituted imidazole derivative (compound 19) [12].These small molecule inhibitors can activate p53 by inhibiting or inducing the dimerization of MDM2 and MDMX at low nanomolar level.However,the limited binding surfaces and poor pharmacological properties hinder the further development of small molecule inhibitors [13].
In recent years,extensive attention has been focused on the application of peptides in drug delivery,biomedicine and clinical therapy,especially for tumor therapy [14–16].Compared with small molecule drugs,peptides offer higher binding affinity and more excellent specificity due to their large interacting surfaces[17,18].As a result,peptides have been considered as effective inhibitors of protein–protein interactions (PPIs) [19].Therefore,it is attractive to inhibit PPIs between p53 and MDM2/MDMX by peptide inhibitors [20].However,although p53 sequence-based linear peptide inhibitors have a high binding affinity for MDM2 and MDMX,their poor proteolytical stability and low intracellular delivery efficiency limit their clinical efficacy [13,21].
The strategy of all-hydrocarbon stapling has been wildly used to improve the protease resistance,membrane permeability and biological activity of peptides [22,23].For example,ATSP-7041,as a stapled peptide,is a highly potent and specific dual inhibitors of MDM2 and MDMX [24].However,the introduction of hydrophobic alkane side chain leads to its poor water solubility [25].Fortunately,the lactam bridges formed by the side chains of aspartate(Asp) and lysine (Lys) are hydrophilic,and thus can improve the aqueous solubility of stapled peptides [26]and induce higherαhelicity than other stapling strategies [27].So,the bicyclic stapling strategy combined with all-hydrocarbon stapling and lactam stapling strategies has better advantages compared with monocyclic stapled peptides [28].
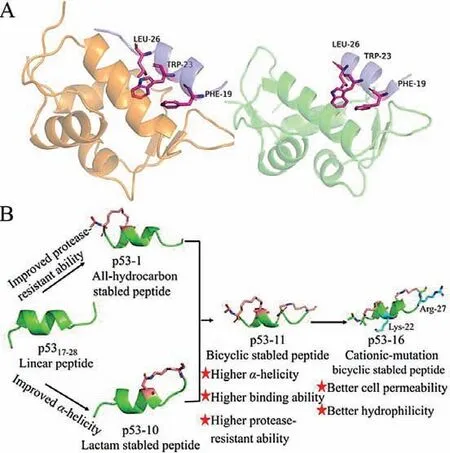
Fig.1.(A) Structure of the binding domain of MDM2/MDMX to p53 binding segment (p53,residues 17–28).Left: p53–MDM2 (blue/yellow) PDB: 3G03;right: p53–MDMX (blue/green) PDB: 2Z5T.(B) Schematic diagram of this research plan.We synthesized stapled peptides p53-1-10 using all-hydrocarbon stapling and lactam stapling strategies to improve protease-resistant ability and α-helicity of p5317–28.The bicyclic stapled peptide p53-11 was synthesized by combining these two strategies,and the cationic-mutation peptide p53-16 based on p53-11 was synthesized to improve cell permeability.
According to X-ray diffraction analysis,the minimal sequence of p53 (p5317–28) binding to MDM2 and MDMX forms an amphiphilicα-helix structure in the complex,which can bind tightly to the hydrophobic cavity of MDM2 (25–129) or MDMX (24–108) [29].Among them,phenylalanine (Phe) 19,tryptophan (Trp) 23 and leucine (Leu) 26 are essential for the interactions of p53 with MDM2 and MDMX [30](Fig.1A).In this study,we maintained the three critical residues in the p53-based peptide design,and synthesized a series of stapled peptides based on p5317–28by using all-hydrocarbon stapling and lactam stapling strategies (Fig.1B).We expected to improve theα-helical stability,protease-resistant ability,membrane permeability and biological activity of p53–MDM2/MDMX inhibitors.
As shown in Table 1,the stapled peptides p53-1-10 were synthesized by replacing the positions at (17,21),(18,22),(20,24),(21,25) and (24,28) of p5317–28by using all-hydrocarbon stapling and lactam stapling strategies.The bicyclic stapled peptides p53-11 and p53-12 were synthesized by combining all-hydrocarbon stapling and lactam stapling strategies.The bicyclic stapled peptides p53-13 and p53-14 were synthesized as control.In addition,we synthesized two cationic-mutation peptides p53-15 (L22K,P27R)and p53-16 (L25K,P27R) to improve cell permeability of bicyclic stapled peptides.The structures of all stapled peptides were shown in Fig.S1 (Supporting information).
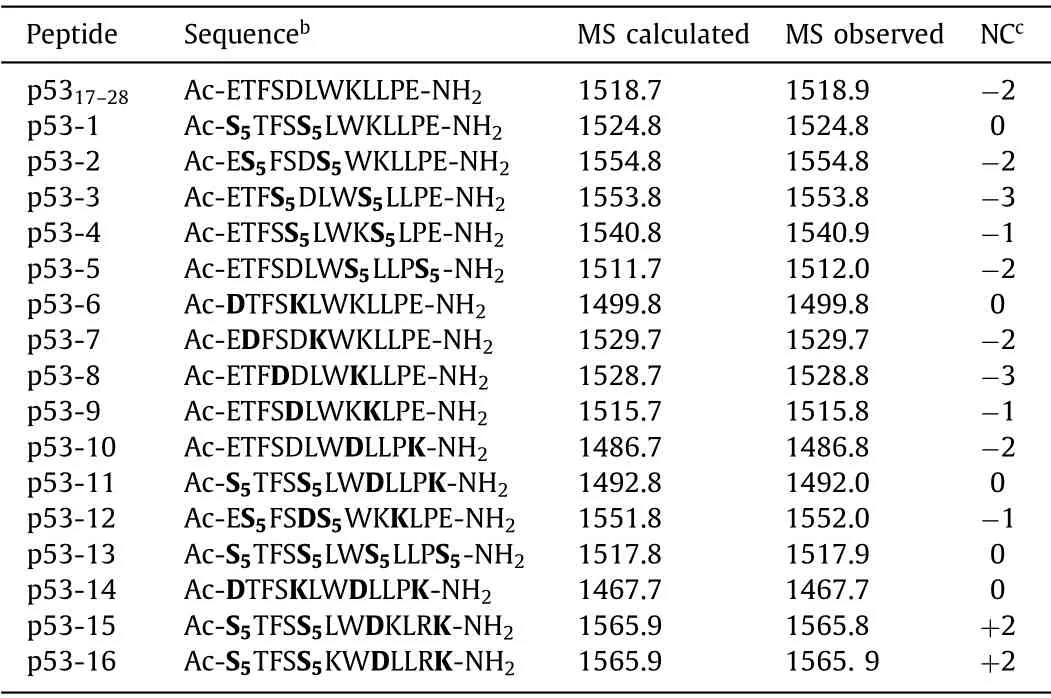
Table 1 Sequences,MS data and net charges of p5317–28 and its analoguesa.
The bicyclic stapled peptide p53-11 was synthesized on Rink Amide MBHA resin using standard Fmoc-based solid phase peptide synthesis (SPPS) as shown in Scheme 1.First,Fmoc-protected amino acids were assembled by using the coupling reagent 5-chloro-1-[bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide hexafluorophosphate (HCTU) andN,N-diisopropyl ethylamine(DIEA).2-(7-Azabenzotriazol-1-yl)-N,N,Nʹ,Nʹ-tetramethyluronium hexafluorophosphate (HATU),1-hydroxy-7-azabenzotriazole(HOAT) and DIEA were used for coupling of (S)-N-Fmoc-2-(4-pentenyl) alanine (S5) [31].Then,the alloc and allyl groups

Scheme 1.Synthetic route of bicyclic stapled peptide p53-11.Conditions: (i) 20% piperidine in N,N-dimethylformamide (DMF) 5 min (2 times),35 °C;(ii) Fmoc–AA–OH (4 equiv.),HCTU (4 equiv.),DIEA (8 equiv.) in N-methylpyrrolidone (NMP),30 min,60 °C;(iii) Fmoc–S5–OH (4 equiv.),HATU (4 equiv.),HOAT (4 equiv.),DIEA (4 equiv.) in NMP,60 min,35 °C;(iv) Pd(PPh3)4 (0.3 equiv.),PhSiH3 (15 equiv.) in DCM,3 h (2 times),25 °C;(v) PyBOP (6 equiv.),DIEA (12 equiv.) in DMF,25 °C,6 h;(vi) 6 mmol/L Grubbs’1st catalyst in DCE,2 h (2 times),35 °C;(vii) Acetic anhydride (4 mL),DIEA (4 mL),10 min (2 times) 35 °C;(viii) Reagent K (82.5% TFA,5% H2O,2.5% ethanedithiol,5%thioanisole and 5% phenol),3 h,35 °C.
were removed with Pd(PPh3)4and PhSiH3.Intramolecular lactamization was performed on the resin using benzotriazol-1-yloxytripyrrolidinophosphonium hexafluoro-phosphate (PyBOP) and DIEA.Next,the ring-closing metathesis (RCM) between two S5was performed with Grubbs’1stcatalyst [32].The N-terminus was acetylated with acetic anhydride and DIEA.Finally,p53-11 was cleaved from the resin by treatment with reagent K [82.5% trifluoroacetic acid (TFA),5% H2O,2.5% ethanedithiol,5% thioanisole and 5% phenol].The crude peptide was further purified by semi-preparative high performance liquid chromatography (HPLC).
The circular dichroism (CD) spectroscopy of p53 analogues in phosphate buffered saline (PBS) showed two negative bands at 208 and 222 nm,and a positive band at 192 nm (Fig.2),which is a typical profile ofα-helicity conformation [33].Stapled peptides p53-1-10 displayed 13%–68%α-helicity in PBS buffer (Table 2),while the p5317–28displayed only 14%α-helicity.It’s worth noting that lactam stapled peptides (p53-6-10) displayed higherα-helicity than all-hydrocarbon stapled peptides (p53-1-5) (Fig.2A),which is consistent with the previously reported that a lactam bridge induced higherα-helicity than other stapling strategies [27,34].Especially,theα-helicity of p53-13 with two hydrocarbon bridges is lower than that of p53-11 with one hydrocarbon and one lactam bridge,and the bicyclic stapled peptide p53-14 with two lactam bridges showed the mostα-helicity (Fig.2B),indicating that the introduction of lactam bridge could improve theα-helicity more effectively.
We further evaluated the binding affinities of p53 analogues with MDM2 and MDMX using surface plasmon resonance (SPR),andKdvalues were shown in Table 2.Compared with linear peptide p5317–28,all-hydrocarbon stapled peptides p53-1 and p53-5 showed higher binding affinity.Especially,theKdvalues of bicyclic stapled peptide p53-11 with MDM2 and MDMX were 3.0 × 10−6mol/L and 1.3 × 10−6mol/L,respectively.It was superior to p53-1 and p53-5,which suggested that stabilizing the peptideα-helical conformation with another lactam bridge could increase its binding affinity for MDM2 and MDMX.Besides,compared with bicyclic all-hydrocarbon stapled peptide p53-13 and lactam stapled peptide p53-14,p53-11 showed higher binding affinity for MDM2 and MDMX.

Table 2 The α-helicity,retention time and Kd values of p5317–28 and its analogues.
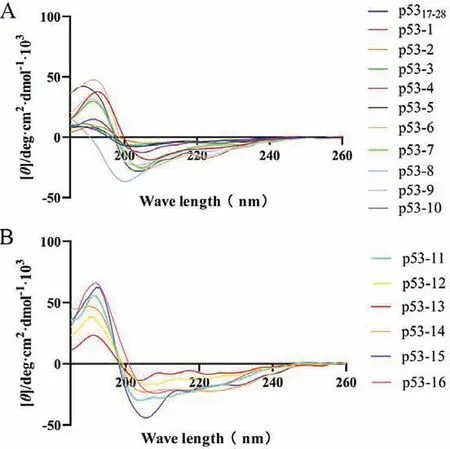
Fig.2.(A) CD spectroscopy of p5317–28 and its monocyclic stapled peptides.(B) CD spectroscopy of bicyclic stapled peptides.
In general,peptides are easy to be degraded by serum proteases and are rapidly cleared in the blood,thus limiting their clinical efficacy [35].So,we assessed the resistance of p53 analogues against protease degradationin vitroat room temperature in PBS buffer(pH 7.4) using a method based on HPLC,following treatment with trypsin.As expected,linear peptide p5317−28was completely degraded in less than 1.5 h after treatment with trypsin,and the half time (t1/2) was 0.49 h.(Fig.3A and Fig.S2 in Supporting information).However,under the same condition,thet1/2of p53-1 and p53-10 were 2.8 h and 1.2 h,respectively,suggesting that all-hydrocarbon stapling and lactam stapling strategies improved protease resistance (Fig.3A).In particular,bicyclic stapled peptide p53-11 showed significantly higher protease resistance,with an 8-fold half time (t1/2=4.2 h) than p5317−28,and even remain more than 12 h (Fig.3A and Fig.S3 in Supporting information).Compared with p53-1 and p53-10,p53-11 showed enhanced protease resistance may because of an additional macrocyclic bridge,whichcould physically block the access of the protease to the cleavable residues [36].
In addition,we explored the stability of p5317−28and p53-11 in human plasma.Similarly,p53-11 showed extremely improved stability than p5317–28(Fig.3B).Linear peptide p5317−28was completely degraded in 6 h,however,p53-11 also remained 68% after 48 h.These results demonstrated that bicyclic stapled peptide effectively improved the stability.
To investigate whether p53 analogs can activate the p53 pathway,we firstly evaluated the cytotoxicity of p53 analogs against human breast tumor cells MCF-7 (express WT p53,MDM2 and MDMX),and human colorectal tumor cells SW480 (carry mutant p53) with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [37].Unfortunately,p53 analogs were incapable of inhibiting the cell activity of MCF-7 cells effectively at a concentration of 20 μmol/L (Fig.S4 in Supporting information)despite all-hydrocarbon stapling and lactam stapling strategies enhanced theα-helicity and binding affinity for MDM2/MDMX of p53.It may due to their inability to cross cell membranes to exert cytotoxicity against tumor cells [22].
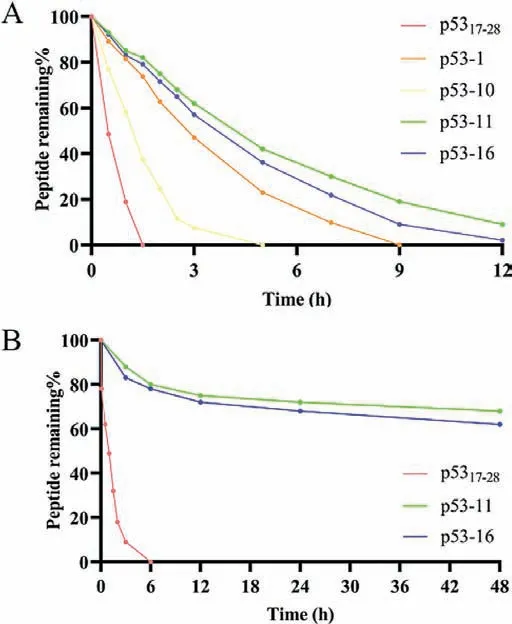
Fig.3.(A) Stability of p5317–28 analogues under trypsin treatment.(B) Stability of p5317–28 analogues in human serum.
To our knowledge,cationicity plays an essential role in the ability of stapled peptides to permeate the cell membranes [38,39].However,p53 analogs carried a net charge of either 0 or −3,which led to little cytotoxicity against tumor cells.Therefore,we selected p53-11 with high binding affinity as the template peptide,and further synthesized two cationic-mutation peptides p53-15 and p53-16 with+2 net charges (Table 1).We observed that cationic-mutation peptide p53-16 reduced MCF-7 cell viability in a dose-dependent manner [half maximal inhibitory concentration(IC50) 17.08 μmol/L]after 72 h incubation (Fig.4A).
We further studied the membrane permeability of p53 analogs using confocal laser scanning microscope (CLSM).CLSM analysis of MCF-7 cells treated with 20 μmol/L fluorescein isothiocyanate(FITC)-labeled p53-16 showed a diffused intracellular localization of the peptide in the cells in different hours (Figs.5A and B),indicating the ability of p53-16 to permeabilize the cell membrane.Besides,we founded that p53-16 significantly entered the cells in 2 h (Fig.S5C in Supporting information).However,p5317–28and p53-11 cannot cross the cell membranes in 4 h (Fig.5A and Figs.S5A and B in Supporting information),which led to their low cytotoxicity.Cytotoxicity of p53-16 was further investigated by repeating the assay using SW480 cells as a negative control,as these cells carried mutant p53 and cannot activate the p53 pathway [40].However,p53-16 cannot significantly reduce SW480 cells viability even at the concentration of 80 μmol/L (Fig.4B).To further determine whether p53-16 can inhibit the interactions of p53–MDM2/MDMX to activate the p53 pathway,we treated MCF-7 and SW480 cells with p53-16 for 24 h and monitored the expression of three p53 target genes,p21,MDM2 and macrophage inhibitory cytokine-1 (MIC-1) by quantitative real-time polymerase chain reaction (qPCR).The expression of mRNA of p21,MDM2 and MIC-1 increased in a dose-dependent phenomenon was only observed in MCF-7,but not in SW480 cells (Fig.4C).These results revealed that p53-16 could permeate cell membrane and selectively inhibit the activity of tumor cellsviaactivating p53 pathway.
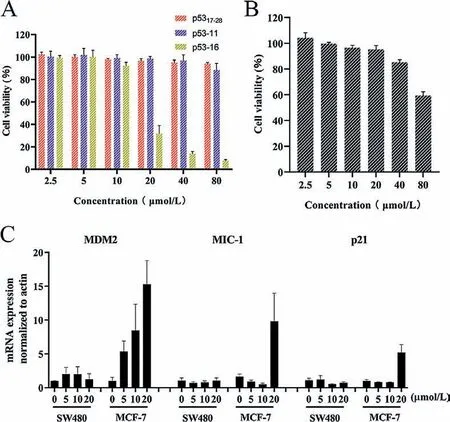
Fig.4.(A) Effect of p5317–28 analogs on the viability of MCF7 cells at different concentrations.(B) Effect of p53-16 on the viability of SW480 cells at different concentrations.(C) Dose-dependent induction of p53 target genes in MCF7 cells 24 h post p53–16 addition.Data presented as mean ± standard deviation (SD),n=3.
Especially,we found theKdof bicyclic stapled peptides p53-16 with MDM2 and MDMX was 2.6 × 10−7mol/L and 1.8 × 10−7mol/L,respectively (Figs.S6A and B in Supporting information),which was significantly superior to p53-1 and p53-5.According to the analysis of the molecular docking (Figs.6A and B),p53-16 displayed a tighter conformation bound to MDM2 and MDMX than that of native peptide p5317–28,with three key residues (Phe-19,Trp-23 and Leu-26) in binding site.In addition,the hydrogen-bond formed between Arg-27 and Tyr-96 will also enhance the binding affinity of p53-16 and MDMX according to previous report [41].Not surprisingly,p53-16 also displayed higherα-helicity than all-hydrocarbon stapled peptides (Fig.2B).And we found the retention time of p53-16 was only 14.9 min (Table 2),it suggested an additional lactam bridge and cationic-mutation strategy improved the hydrophilicity of p53-16.
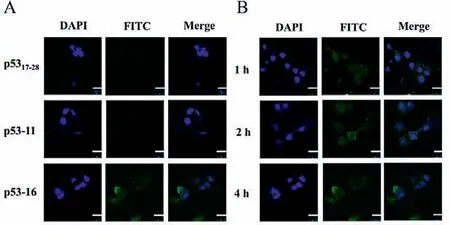
Fig.5.(A) CLSM of FITC-labeled p5317–28 analogs (20 μmol/L) localization in MCF-7 cells in vitro for 4 h.The nuclei were stained with DAPI (blue).(B) CLSM images of FITC-labeled p53-16 (20 μmol/L) localization in MCF-7 cells in vitro for 1,2 and 4 h,respectively.The scale bar is 25 μm.
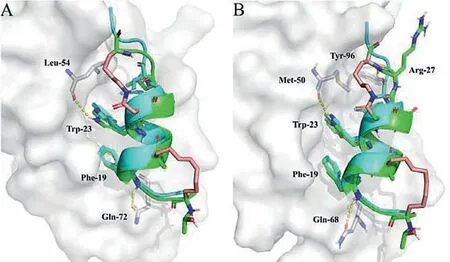
Fig.6.Structure of p5317–28 or p53-16 in complex with MDM2 (A) and MDMX (B).The MDM2 and MDMX were shown in the surface representation (gray).p53-16(green) was aligned with p5317–28 (cyan).Interacted residues were shown as stick models.Hydrogen-bonds were shown as yellow dashed line.
In conclusion,we synthesized the all-hydrocarbon stapled peptides and lactam stapled peptides based on p53,respectively.We found that the C-terminal lactam bridge strategy induced moreα-helicity,and the N-terminal hydrocarbon strategy improved the protease resistance effectively.Hence,we synthesized the bicyclic stapled peptides p53-11 by combining the all-hydrocarbon stapling strategy and lactam stapling strategies.p53-11 significantly improved theα-helicity,protease resistance,stability in human plasma and binding affinity for MDM2 and MDMX.Especially,the cationic-mutation bicyclic stapled peptide p53-16 showed nanomolar binding affinity for MDM2 and MDMX,could penetrate the cell membrane and selectively inhibit the activity of tumor cellsviaactivating p53 pathwayin vitro.As a result,p53-16 was proved as a potential dual inhibitor for the interactions with MDM2 and MDMX.This is the first time that the bicyclic stapling strategy has been applied to the design and synthesis of inhibitors of p53 interactions with MDM2 and MDMX,and it is a promising drug design strategy for PPIs inhibitors.
Declaration of competing interest
There are no conflicts to declare.
Acknowledgments
This research was supported by National Natural Science Foundation of China-Shandong Joint Fund (No.U1606403) and Innovation Project of Qingdao National Laboratory for Marine Science and Technology (No.2015ASKJ02).We are grateful to the Instrumental Analysis Center of Ocean University of China for mass spectrometric analysis.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.08.130.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis