
Application of Langlois’reagent (NaSO2CF3) in C–H functionalisation
2022-06-18 10:52JiabinShenJunXuLeiHeChenfengLiangWanmeiLi
Chinese Chemical Letters 2022年3期
Jiabin Shen,Jun Xu,Lei He,Chenfeng Liang,Wanmei Li
College of Material,Chemistry and Chemical Engineering,Key Laboratory of Organosilicon Chemistry and Material Technology,Ministry of Education,Hangzhou Normal University,Hangzhou 311121,China
Keywords:Trifluoromethylation Langlois’reagent Reaction mechanism Synthetic method C–H activation
ABSTRACT The process of selectively introducing a CF3 group into an organic molecule using inexpensive,stable,and solid sodium trifluoromethanesulfinate has rapidly advanced in recent years to become an eco-friendly method used by organic chemists to synthesize various natural and bioactive molecules.This review focuses on advances made within the last five years regarding C–H functionalisation,namely thermochemical C(sp2)–H (thio)trifluoromethylations,photochemical C(sp2)–H trifluoromethylations,and electrochemical C(sp2)–H trifluoromethylations,using Langlois’reagent (NaSO2CF3).
1.Introduction
Due to its small atomic radius and strong electronegativity,the introduction of atomic fluorine often engenders compounds with unique physical,chemical,biological,and functional properties [1–7].Among them,trifluoromethyl-substituted organic compounds have found important applications as pharmaceuticals,agrochemicals,and organic functional materials because the biological and functional activities of organic compounds can be significantly influenced by the trifluoromethyl group [8–11].Therefore,a number of trifluoromethyl-containing compounds have been developed as efficient pharmaceuticals and agrochemicals over the years [12–15].Swarts-type reactions are traditional methods for incorporating CF3groups into aromatic backbones;however,these reactions require chlorine–fluorine exchange in the presence of HF under harsh conditions,and their range of tolerated substrates is relatively narrow [16,17].Such processes are not appropriate for the late-stage incorporation of a CF3group into aromatic structures.To overcome these shortcomings,methods that directly trifluoromethylate aromatic compounds have been developed,including the use of aryl-boronic acids,aryl-fluoroborates,and arenediazonium salts [18–24].Although significant advances have been made in the trifluoromethylation field,the flexibility,eco-friendliness,atom-economy,and selectivity of trifluoromethylation chemistry requires further improvement.For example,the selective introduction of a CF3group into an organic molecule through C–H functionalisation has attracted immense attention in recent years [25–28].Tomashenko and Grushin reviewed and analysed aromatic C–CF3bond-forming reactions using metals and their complexes in 2011,and provided an outlook for future studies with emphasis on practical features [29].Whereafter,Studer summarized recent developments in radical trifluoromethylation reactions and promoted the development of new methods for radical trifluoromethylation in 2012 [30].Zhang’s group published a review about the application of Langlois’reagent (NaSO2CF3) in trifluoromethylation reactions over the period of 2011 to 2014,which promoted the development of new methods for trifluoromethylations employing this reagent [31].Subsequently,numerous papers have appeared reporting the use of NaSO2CF3in C–H functionalisation.Therefore,it is necessary to summarize developments made from 2015 to the present regarding the application of Langlois’reagent in C–H functionalisation.
The Langlois’ reagent (NaSO2CF3),which provides an electrophilic trifluoromethyl radical (CF3˙) to electron-rich double bonds and arenes,was first reported in the 1980s by Langlois and co-workers [32,33].Since then,a considerable number of papers have reported the use of NaSO2CF3for the formation of C–CF3bonds.Compared with other CF3sources,such as the Togni,Umemoto-Yagopulskii and Umemoto reagents,Langlois’reagent is more stable,commercially available,and less expensive.Langlois’reagent is also obtained in yields ranging from 74% to quantitative when trifluoromethylated sulfoxide is treated with sodium methoxide in methanol (Schemes 1a and b),or when CF3Cl is reacted with Na2S2O4in DMSO (Scheme 1c) [34,35].
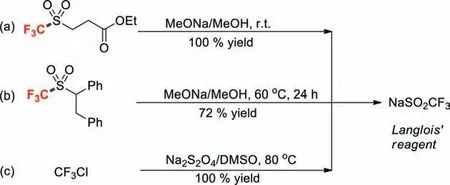
Scheme 1.Syntheses of Langlois’reagent.
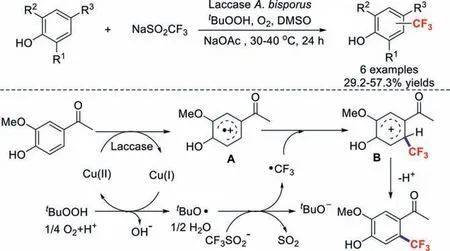
Scheme 2.Possible reaction mechanism for the reaction of an unprotected phenol with NaSO2CF3.
This paper reviews the use of NaSO2CF3in C–H functionalisation chemistry since 2015.The results are discussed and classified according to the catalytic type.Particular attention is devoted to representative reaction mechanisms.Selected examples involving typical substrates are displayed as well.In addition,we summarise current research progress in this field and discuss future expectations.
2.Application of Langlois’reagent to C–H functionalization
C−H functionalization reactions have great potential for streamlining the syntheses of complex molecules and are currently “hot”topics in the field of organic synthesis.Herein,this approach is summarised in the following three sections: (i) thermochemical C(sp2)–H (thio)trifluoromethylations,(ii) photochemical C(sp2)–H trifluoromethylations,and (iii) electrochemical C(sp2)–H trifluoromethylations.
2.1.Thermochemical C(sp2)–H (thio)trifluoromethylations
In contrast to the common strategy used for the introduction of the CF3group into an aromatic compound (metalmediated/catalysed functional-group interconversion),Simonet al.reported an efficient and selective method for the trifluoromethylation of an unprotected phenol through the biocatalytic introduction of a trifluoromethyl group derived from a common precursor [36].The reaction proceeds under mild conditions with high regioselectivity,a wide substrate range,and good functional-group compatibility.A mechanism for the reaction of an unprotected phenol with NaSO2CF3is proposed in Scheme 2.First,the substrate is oxidised to radical cation A by laccase and Cu(II);meanwhile,the CF3˙radical is formed by the reaction of TBHP with Cu(I).Subsequent freeradical coupling leads to intermediate B,which undergoes deprotonation to afford the products.
In 2018,our group developed the practical and economical Cucatalysed picolinamide-directed C–H trifluoromethylation of aniline at itsortho-position (Scheme 3).This conversion has a broad substrate scope and the products were obtained in good yields.More importantly,this trifluoromethylation reaction was used for the efficient synthesis of floctafenine [37].
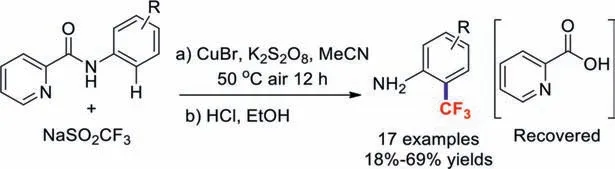
Scheme 3.Cu-catalysed trifluoromethylations at the ortho positions of anilines.
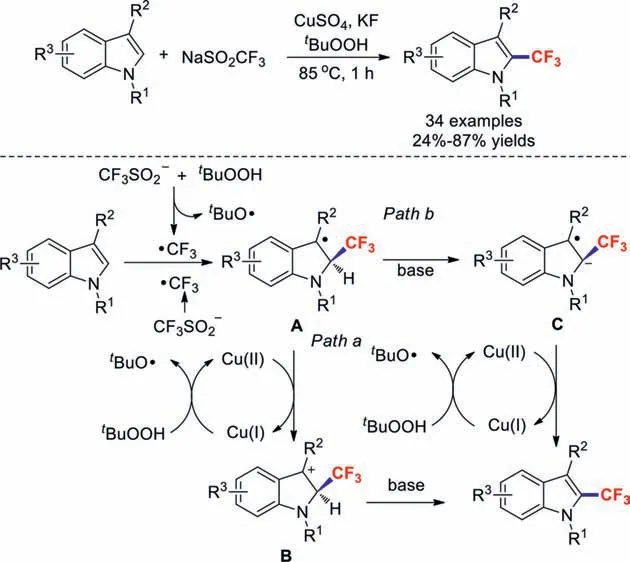
Scheme 4.Possible reaction mechanisms of CuSO4-catalysed oxidative trifluoromethylations affording novel C2-trifluoromethylated indole moieties.
In 2019,the research team of Shi used indoles as substrates,tBuOOH as an oxidant,and KF as a base in CuSO4-catalysed oxidative trifluoromethylations to afford novel C2-trifluoromethylated indole moieties [38].It is worth noting that the Cu(II)/Cu(I) redox process is indispensable in this reaction based on control experiments (Scheme 4).Initially,the CF3˙radical is generated through oxidation bytBuOOH.Alternatively,CF3˙is formed from CF3SO2−andtBuO˙.The indole is then attacked by CF3˙to produce intermediate A,which is then oxidised to intermediate B in the presence of Cu(II).The target product is finally obtained by deprotonation.In addition,Cu(I) is oxidised bytBuOOH to Cu(II) to complete the catalytic cycle (path a,Scheme 4).Considering the alkaline reaction conditions and that theα-CF3–H proton would be highly acidic,we proposed another possible reaction mechanism (path b,Scheme 4).Intermediate A could conduct a deprotonation to form radical anion C in the presence of base.Then,the final product could be afforded through single electron oxidation.
Recently,the development of methods for introducing trifluoromethyl groups into alkenes has received considerable attention among the synthesis community.In early 2017,Wuet al.demonstrated efficient copper(I)-catalysed trifluoromethylation of styrene derivatives in the presence oftBuOOtBu [39].A wide range of functional groups,including halogens,ethers,thioethers,amines,and thiophenes,were found to be compatible under the standard reaction conditions (Scheme 5).
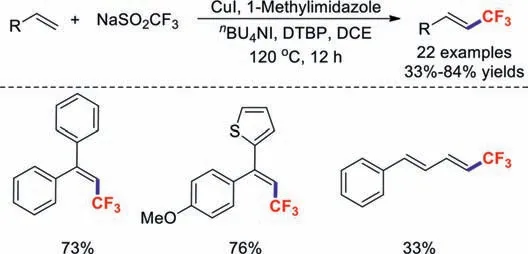
Scheme 5.Trifluoromethylations of styrenes.
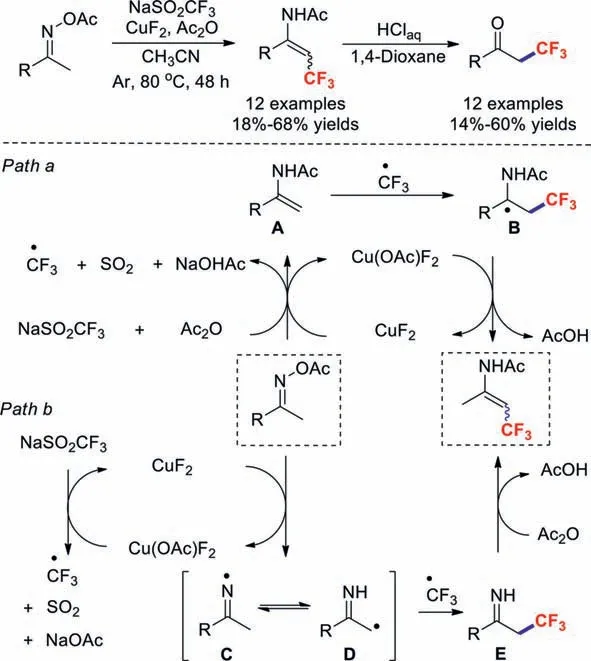
Scheme 6.Trifluoromethylations of oxime acetates.
Meanwhile,Selander’s group disclosed a simple and efficient copper(II)-catalysed trifluoromethylation reaction for oxime acetates in the absence of an external oxidant [40].Due to the polarised N–O bond,which is reductively cleavable by transition metals,this transformation featured mild conditions and a broad substrate scope.Two possible mechanistic pathways were envisaged for this reaction based on control experiments: A radical/enamide addition pathway (path a,Scheme 6),and a radical/radical coupling pathway (path b,Scheme 6).In path a,oxime acetate is reduced to enamide A in the presence of CuF2,NaSO2CF3and Ac2O.Meanwhile,a CF3radical is generatedviathe same process.Subsequently,thein situ-generated CF3radical adds to enamide A,affording intermediate B.Thereafter,the trifluoromethylated enamide is formed by the oxidation of the Cu(III) catalyst.Alternatively,inpath b,oxime acetate is reduced by CuF2to give imine radical C.Upon the isomerization of C and D followed by coupling with CF3radicals and acylation,the product is formed.
In 2016,our group developed a practical and economical chitosan@Cu(OAc)2-catalysed method for remote trifluoromethylations of aminoquinolines [41].This C–C coupling reaction was highly efficient and tolerated a wide variety of functional groups.More importantly,the copper catalyst could be reused five times without significant loss of catalytic activity (Scheme 7).
Compared to the rapid development of copper-promoted trifluoromethylation chemistry [42–46],the development of other metal-catalysed trifluoromethylation reactions has been relatively slow.In 2017,our team reported the nickel(II)-promoted trifluoromethylations of arylamine derivatives at theirorthopositions in water through a coordinating activation strategy [47].Furthermore,this transformation method was successfully used to effi-ciently synthesize acid red 266 (Scheme 8).
In 2017,Mizuno and co-workers disclosed the first protocol for a direct C–H trifluoromethylation involving the catalysis of a (hetero)arene with a vanadium-containing heteropoly acid [48].Various structurally diverse (hetero)arenes were converted into the corresponding trifluoromethylated productsviaradical pathways(Scheme 9).
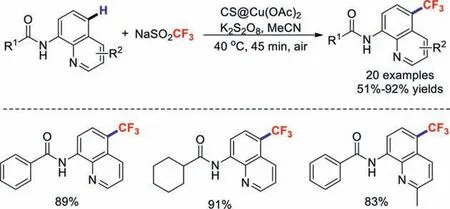
Scheme 7.Chitosan@Cu(OAc)2-catalysed remote trifluoromethylations.
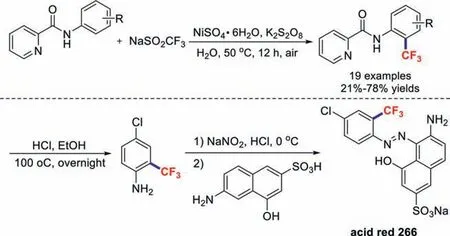
Scheme 8.Nickel(II)-promoted trifluoromethylation reactions.
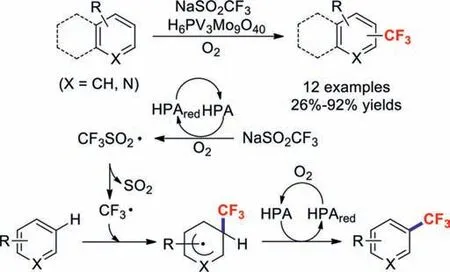
Scheme 9.Phosphovanadomolybdic acid catalysed trifluoromethylation reactions with heteroarenes.
Most trifluoromethylation reaction systems use transition-metal catalysts,which are costly,toxic,and often found as undesirable residual impurities in pharmaceutical products.Hence,it is important to establish economical and transition-metal-free trifluoromethylation methods [49–54].In 2016,a mild and practical catalyst-free strategy for the C–H trifluoromethylations of arenes was described by the research group of Gong [55].This transformation possesses the advantages of operational simplicity,mild reaction conditions,environmental friendliness,and water compatibility.A reasonable mechanism was proposed (Scheme 10),which includes radical initiation followed by radical addition and the loss of a hydrogen atom during aromatisation.The proposed mechanism shows that the alkoxy group plays an important role in this transformation because it stabilizes the free-radical intermediate.
Recently,hypervalent iodine oxidants have received extensive attention due to their low toxicities and high reactivities [56,57].Wuet al.developed an oxidative trifluoromethylation procedure for aminoquinoline amides that uses NaSO2CF3and PIDA under metal-free conditions in 2016 [58].In this method,the remote C–H trifluoromethylation of 8-aminoquinoline occurs at the C5 position.Based on a control experiment and literature precedence,a plausible mechanism was proposed (Scheme 11).Firstly,the trifluoromethyl radical is generated from NaSO2CF3in the presence of PIDA,which then selectively adds to the C5 position of the substrate to form intermediate radical A.Subsequent oxidation with PIDA forms carbocation B,which undergoes deprotonation to give the desired product.
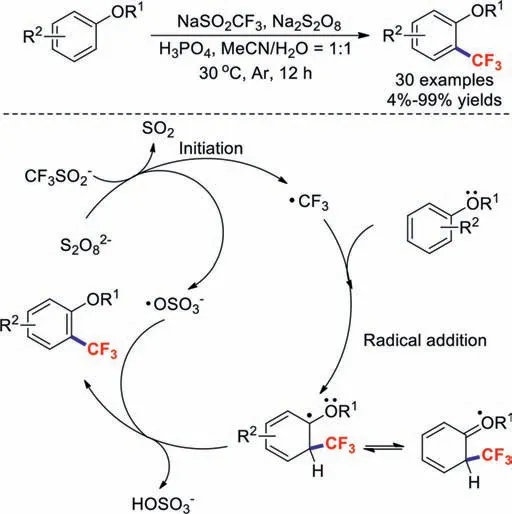
Scheme 10.Possible reaction mechanism for the C–H trifluoromethylations of arenes.
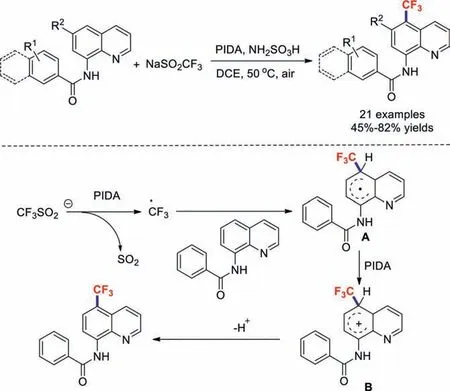
Scheme 11.Possible reaction mechanism of a metal-free NaSO2CF3-based oxidative trifluoromethylation procedure for aminoquinoline amides.
Xu and co-workers in 2017 reported a simple and effi-cient protocol for the preparation of tri-/difluoromethylatedN,Ndialkylhydrazones [59].PhI(OAc)2was used as the oxidant to generate CF3˙from NaSO2CF3with NaHCO3acting as the base.A series of functional groups,including halides,nitriles,nitro moieties,and esters,were well tolerated.It should be noted that heterocyclic hydrazones are also compatible with these mild reaction conditions.The corresponding products were obtained in excellent yields (Scheme 12).Almost concurrently,the research group of Feng demonstrated a metal- and base-free method for the synthesis of trifluoromethylatedN,N-disubstituted hydrazones that was also cost-effective [60].Based on a control experiment and literature precedence,the authors provided a possible mechanism for this transformation.Initially,intermediate A is formed by the reaction of NaSO2CF3with PhI(OAc)2,which produces two free radicals,B and CF3˙,and then attacks the hydrazone to form the trifluoromethylated aminyl radical.The final product is obtained through oxidation of this intermediate followed by proton abstraction by acetate derived from radical B.
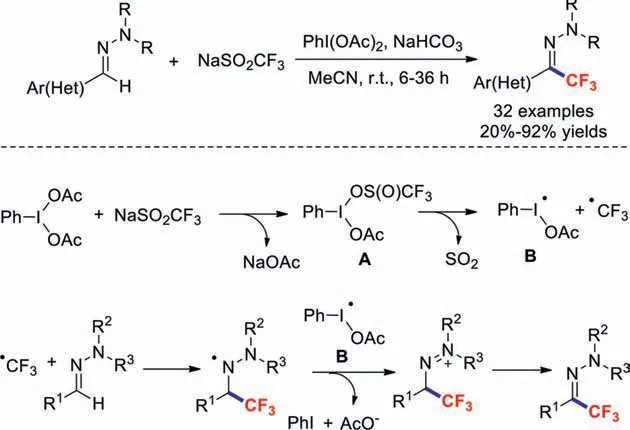
Scheme 12.Trifluoromethylations of N,N-disubstituted hydrazones.
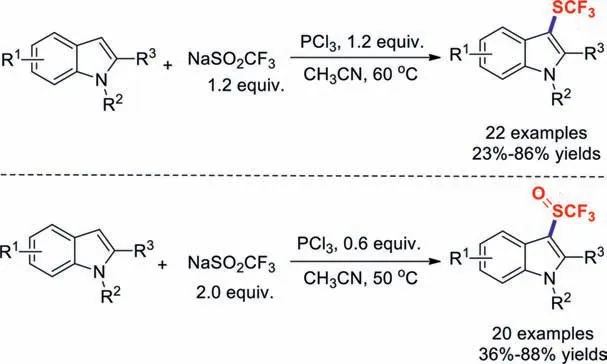
Scheme 13.Trifluoromethylthiolations of indole derivatives.
Trifluoromethyl sulphides (CF3SR) play important roles and are potentially useful reagents in organofluorine chemistry because they exhibit electron-withdrawing effects and high lipophilicity.The Langlois’reagent,known to act as a trifluoromethyl radical source,can generate SCF3in the absence of any additional sulphur sources;however,an additional phosphor source is indispensable in this process [61–66].In 2017,transition-metal-free direct trifluoromethylthiolations of indole derivatives with NaSO2CF3in the presence of PCl3were developed by Lu and co-workers(Scheme 13).Other electron-rich aromatics were also obtained in moderate yields.It is worth mentioning that controlling the dosages of NaSO2CF3and PCl3can result in the selective formation of either the trifluoromethylthiolated or trifluoromethylsulphinylated products in good yields [67].
2.2.Photochemical C(sp2)–H trifluoromethylations
Photocatalytic radical-mediated trifluoromethylation chemistry provides an attractive platform for modifying various biologically important and functionalised (hetero)cycles [68,69].In 2016,the Rueping group developed 4,4′-dimethoxybenzophenone as an innovative,efficient,and mild photoredox catalyst for promoting the trifluoromethylations of (hetero)aromatics and olefins [70].4,4-Dimethoxybenzophenone was excited by 350 nm light and oxidized NaSO2CF3to form a trifluoromethyl radical (Scheme 14).In 2017,a similar reaction was reported by Ravelli and co-workers,which used tetrabutylammonium decatungstate (TBADT) as the photoredox catalyst [71](Scheme 15).This method features good regioselectivity,a wide substrate scope,and excellent functionalgroup tolerance;it also provides an alternative approach for the synthesis of fluorine-containing molecules.Based on experimental evidence,the key to this reaction lies with the strong oxidising properties of the excited decatungstate anion.
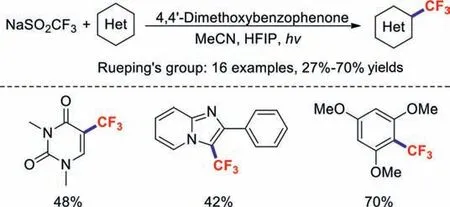
Scheme 14.Photoorganocatalytic trifluoromethylations of (hetero)aromatics.
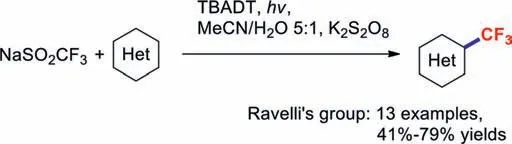
Scheme 15.Photocatalytic radical-mediated trifluoromethylation reactions involving (hetero)cycles.

Scheme 16.Representative examples of trifluoromethylation reactions.
In 2018,the research team of Leadbeater discovered a simple and facile method for the direct introduction of the trifluoromethyl group to indole scaffolds using cheap and commercially available anthraquinone-2-carboxylic acid (AQN-2COOH) as the photoredox catalyst [72].The target products were in full accordance with the results of the computed reaction kinetics.Soon afterwards,Zhang’s group also reported an efficient and mild method for the trifluoromethylation of imidazopyridines using the same photoredox catalyst (AQN-2COOH).This convenient and efficient reaction was proved to be useful for the preparation of a range of substituted imidazopyridines [73].Using methylene blue as the photoredox catalyst,the novel photoredox-catalysed regioselective C3–H trifluoromethylation of 2H-indazoles was subsequently demonstrated by Muruganet al.[74].The reaction conditions of those transformations are summarised in Scheme 16.
The quinoxalinone motif is an important type of organic compound due to its widespread distribution in natural products and pharmaceuticals.Weiet al.reported a practical method for the C–H trifluoromethylation of quinoxalinones using Eosin Y as the photocatalyst and blue LEDs as the light source [75].Unlike previous procedures,these transformations afforded the corresponding products in moderate-to-good yields in air and under additive-free conditions (Scheme 17).Control experiments revealed that a radical mechanism is responsible for this reaction (Path a,Scheme 17).Excited Eosin Y∗is first generated by visible light,which then reacts with NaSO2CF3in a SET process to generate CF3˙.The trifluoromethyl radical further attacks the substrate to form intermediate A;a 1,2-hydrogen shift then generates intermediate B.Meanwhile,another SET process between Eosin Y∗and O2provides an O2∗species along with the regeneration of Eosin Y.Intermediate B is subsequently transformed into intermediate C,which is oxidised by O22−∗to afford the target product.Similarly,we propose another possible mechanism for this reaction (Path b,Scheme 17).Intermediate A would undergo deprotonation to form radical anion E,which is then oxidised by O2−∗to afford the target product.
Organic semiconductor materials are photo and chemically stable relative to other reactive radicals and nucleophiles and possess convenient bandgaps between their valence band maxima and conduction band minima that facilitate the control of oxidations and reductions of numerous practical substrates [76,77].In 2019 Ghosh and co-workers reported the use of the organic semiconductor mesoporous graphitic carbon nitride (mpg-CN) as a heterogeneous photoredox catalyst for C–H trifluoromethylations of arenes and (hetero)arenes [78].This novel approach is markedly different from those employing traditional photocatalysts,which typically react with single reactants either through oxidation or reduction.In the method of Königet al.,the organic semiconductor mpg-CN,upon photoexcitation,naturally generates electron–hole pairs on the catalyst surface,similar to primary batteries.Consequently,oxidative and reductive redox steps yield arene products functionalized at two distinct C–H sites.This approach can be potentially used to specifically form the C–CF3bond (Scheme 18).
In 2018,Xia and co-workers described Fe-catalysedorthotrifluoromethylations of anilines under ultraviolet irradiation [79].This effective reaction overcomes the shortcomings of our reported method in which substrates with electron-withdrawing groups were incompatible.It should be noted that acetone essentially acts as the solvent and a low-cost radical initiator.A possible mechanism is shown in Scheme 19.First,FeIILn is oxidized by air to afford FeIIILn,which then combines with the substrate to generate intermediate A.Next,SET results in intermediate B.Meanwhile,a trifluoromethyl radical is generated from NaSO2CF3under ultraviolet irradiation,which attacks intermediate B to produce intermediate C.Subsequently,intermediate C is transformed into cationic intermediate D through intramolecular trifluoromethylationviaa pentacyclic transition state.Through deprotonation and metal dissociation process,the target product is obtained.
Soon afterwards,the research team of Li reported a similar visible-light mediatedortho-C−H trifluoromethylation of aniline in the presence of CuCl2[80].In this transformation,in addition to picolinamides,other aromatic amides,alkyl anilides,and lactams also readily afforded the corresponding products (Scheme 20).
Recently,visible-light-promoted organic transformation reactions without photocatalysts have received considerable attention[81–83].In 2016,Li and co-workers reported a simple,metal- and oxidant-free photochemical strategy for the trifluoromethylations of arenes and (hetero)arenes (Scheme 21).Significantly,aliphatic ketones act as promising radical initiators in this photochemical strategy upon their irradiation with light.Meanwhile,this efficient and practical approach provides a green route for the cost-effective large-scale synthesis of trifluoromethylated chemicals [84].
Two years later,the Li group revealed a simple,metal- and oxidant-free photocatalysis strategy for the light-mediated trifluoromethylation of coumarins [85].This photochemical strategy also provides an environmentally conscious route for the efficient largescale synthesis of trifluoromethylated chemicals.Later,the research team of Jin also developed a novel and regioselective method for trifluoromethylations of quinoxalin-2(1H)-ones [86].This transformation features mild conditions,good regioselectivity,a broad substrate scope,and moderate-to-good product yields (Scheme 22).
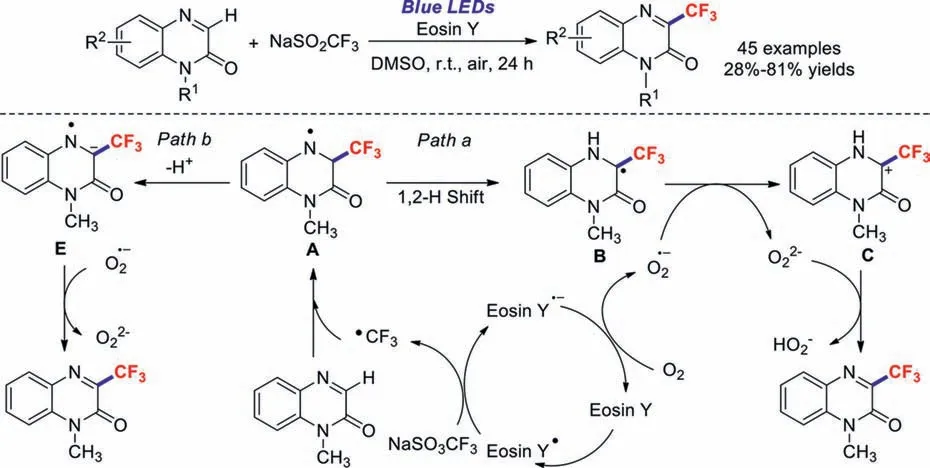
Scheme 17.Possible reaction mechanism for the C–H trifluoromethylation of quinoxalinones using Eosin Y as the photocatalyst and blue LEDs.

Scheme 18.Selected examples of mpg-CN-catalysed trifluoromethylation reactions.
As part of their continued interest in visible-light-induced organic reactions in the absence of an external photosensitiser,the Wang group described the highly selective remote C–H trifluoromethylation of 8-aminoquinoline under visible-light irradiation[87].These photocatalyst-free,simple,and mild conditions generated various 5-trifluoromethylated quinolines in good yields.Soon afterwards,they demonstrated the visible-light-induced Pdcatalysedortho-trifluoromethylations of acetanilides [88];this protocol featured good functional-group tolerance,a wide substrate scope,and operational simplicity,and provided the desired products in moderate-to-good yields (Scheme 23).
It is worth mentioning that a safe,efficient,and sustainable continuous-flow trifluoromethylation strategy for arenes and (hetero)arenes was reported by the research group of Noёl [89].A series of substrates of high interest to drug discovery programs were trifluoromethylated in moderate-to-good yields (Scheme 24).
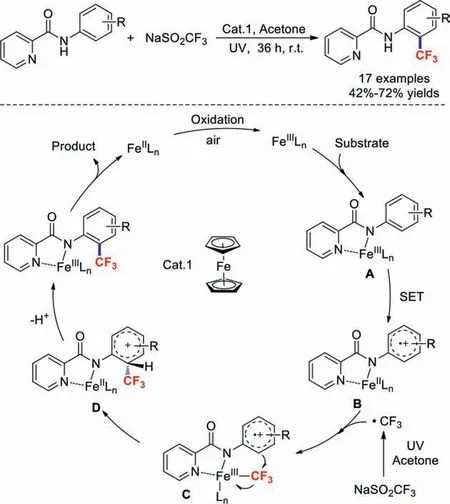
Scheme 19.Iron-catalysed ortho trifluoromethylation of anilines.
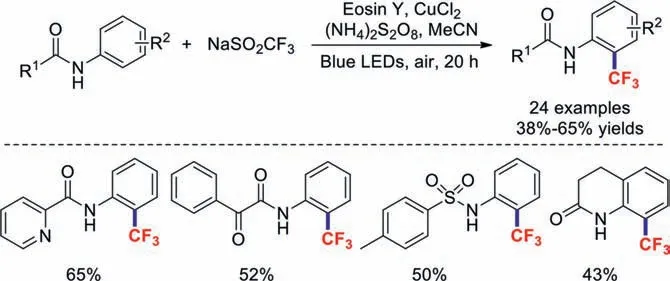
Scheme 20.Visible-light-mediated ortho trifluoromethylation of anilines.
Most photoredox reactions efficiently proceed under mild and simple operational conditions.The photocatalyst in a photocatalytic cycle is usually stimulated to its excited state in the presence of visible light.In photocatalytic reactions involving NaSO2CF3,this excited state then provides a CF3radical from NaSO2CF3viaSET.Subsequent radical additions to unsaturated bonds with further transformations produce the corresponding fluorinated products.The significant advantages of photocatalysed trifluoromethylation chemistry,including mild reaction conditions,good functionalgroup tolerance,and experimental ease,make such approaches attractive for further industrial applications.
2.3.Electrochemical C(sp2)–H trifluoromethylations
Due to its high selectivity,relatively mild conditions,and substrate tolerance,electroorganic synthesis has been expanded over many years to include various transformations [90–93].In 2019,Deng and co-workers disclosed the mild and reagentfree electrochemical oxidative radical C–H trifluoromethylation of arenes/heteroarenes [94].It should be noted that this reaction could be conducted on the gram scale with high reaction efficiency at a current of 20 mA over 36 h (Scheme 25).
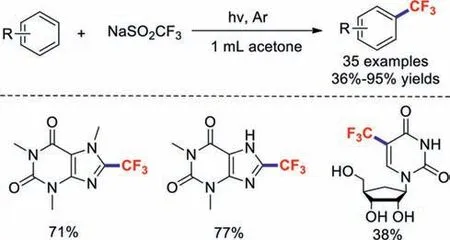
Scheme 21.Representative examples of trifluoromethylation reactions.
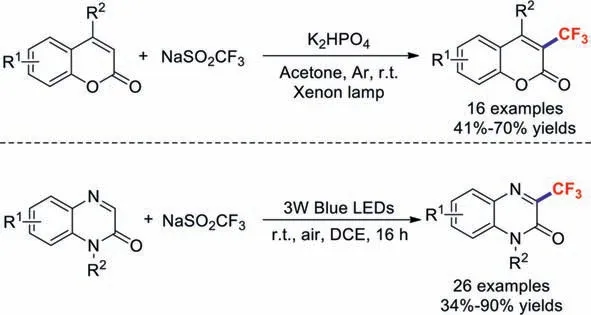
Scheme 22.Light-mediated trifluoromethylations.
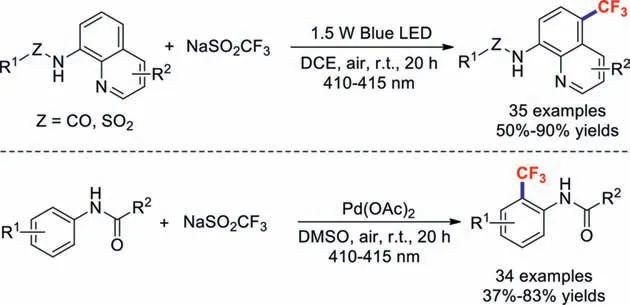
Scheme 23.Visible-light-induced remote C–H trifluoromethylations.
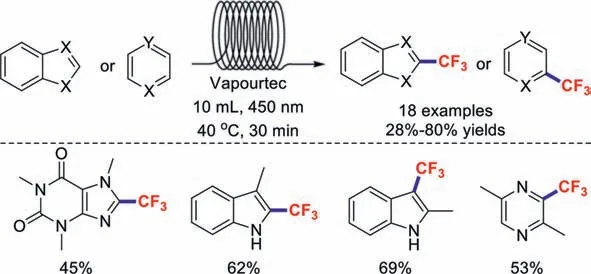
Scheme 24.Representative examples of continuous-flow trifluoromethylation reactions.
In 2020,the research team of Ackermann discovered the unexpected electrophotochemical C–H trifluoromethylation of(Het)arenes [95].This strategy avoided the use of expensive and toxic chemical oxidants,and provided synthetically useful bioactive molecules,including caffeine,pentoxifylline,doxofylline,theobromine,methyl estrone,and tryptophan derivatives,in high yields.Based on mechanistic findings,a plausible mechanism is proposed in Scheme 26.First,an excited state,Mes-Acr+∗,is generated under the irradiation of visible light.Next,SET occurs between Mes-Acr+∗and CF3SO2−to form a trifluoromethyl radical and Mes-Acr∗.Then,the trifluoromethyl radical attacks the substrate to produce intermediate A.After that,intermediate A can be transformed into cationic Wheland complex B through SET or anodic oxidation.Finally,proton abstraction delivers the desired product.In the meantime,protons are reduced to generate H2at the cathode.
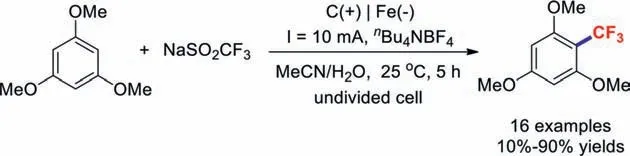
Scheme 25.Electrochemical oxidative trifluoromethylations of arenes.
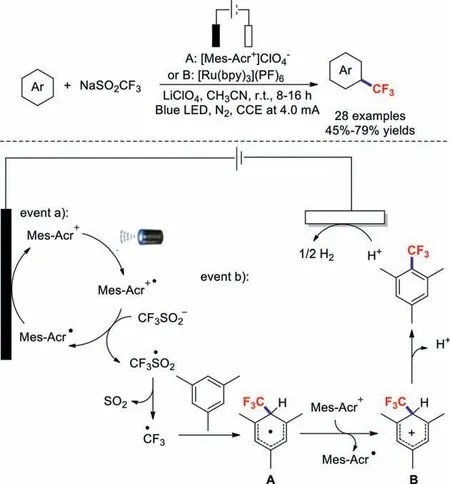
Scheme 26.Electrophotochemical C-H trifluoromethylations of (Het)arenes.
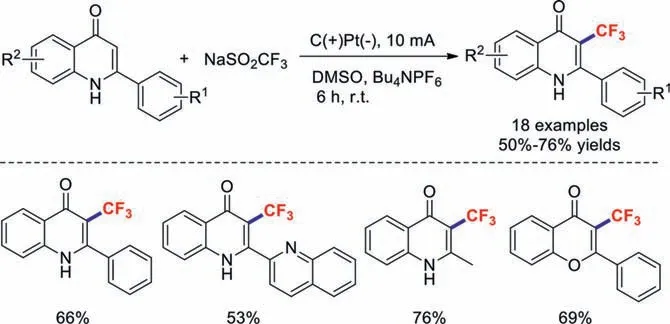
Scheme 27.Electrochemical trifluoromethylations of quinolinones.
Soon afterwards,Chu and co-workers directly electrochemically trifluoromethylated quinolinones using NaSO2CF3as the trifluoromethyl source [96].This esterification protocol gave the corresponding products in moderate-to-good yields under metal-free and oxidant-free conditions (Scheme 27).
3.Summary and outlook
Herein,we overviewed recent advances (over the past five years) in the use of Langlois’Reagent (NaSO2CF3) in C–H functionalisation chemistry.three types of transformations were discussed: (i) thermochemical C(sp2)–H trifluoromethylations,(ii)photochemical C(sp2)–H trifluoromethylations,and (iii) electrochemical C(sp2)–H trifluoromethylations.Although considerable effort has been directed towards developing C–H trifluoromethylation chemistry,which has resulted in great advances,additional research is still required for the further advancement of this field.For example,C(sp3)–H trifluoromethylations of alkanes using NaSO2CF3as the trifluoromethylating reagent have not yet been reported.Research into such a transformation is still relatively new,making it a prospective research area.Furthermore,to date,there are no reports on asymmetric C–H trifluoromethylation reactions using NaSO2CF3as the trifluoromethylating reagent.These areas need to be further explored.In addition,we discussed some suggested mechanisms and proposed other possible mechanisms for the reviewed transformations.According to previous reports [97,98],when a CF3radical adds to a substrate and generates a trifluoromethylated radical,there are two possible routes for the generation of the final product (Scheme 28),depending on the reaction conditions.One of the transformations involves the trifluoromethylated radical undergoing an electron-transfer (ET) process to form a carbocation,which then undergoes a proton-transfer(PT) process to give the final product.For the other transformation,theα-CF3–H proton needs to be highly acidic,so that the trifluoromethylated radical would undergo a PT process to give the corresponding carbanion in the presence of base.The carbanion would then go through an ET process to give the product.Overall,we hope that these novel methods can be used in industrial processes towards chemically valuable organic compounds and believe that new methods for the formation of C–CF3bonds using the Langlois’reagent will continue to be developed.
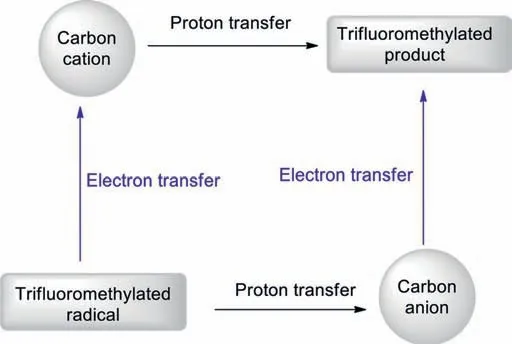
Scheme 28.Two possible mechanisms for the generation of products from trifluoromethylated radicals.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We thank the Natural Science Foundation of Zhejiang Province(No.LY21B060009) for financial support.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis