
Transition metal-catalyzed conversion of aldehydes to ketones
2022-06-18 10:52ZijunYnPnLinShoQingQingFeipengLiuXuchoWngYongjieLiZiQingRong
Chinese Chemical Letters 2022年3期
Zijun Yn,Pn-Lin Sho,Qing Qing,Feipeng Liu,Xucho Wng,Yongjie Li,Zi-Qing Rong
a Frontiers Science Center for Flexible Electronics (FSCFE),Shaanxi Institute of Flexible Electronics (SIFE) &Shaanxi Institute of Biomedical Materials and Engineering (SIBME),Northwestern Polytechnical University (NPU),Xi’an 710072,China
b College of Innovation and Entrepreneurship,Southern University of Science and Technology,Shenzhen 518055,China
Keywords:Homogeneous catalysis Transition metal catalysis Direct conversion Ketone synthesis C-H bond functionalization
ABSTRACT Ketones are one of the most important classes of organic compounds,and widely present in various pharmacological compounds,biologically active molecules and functional materials.Over the past few decades,transition metal-catalyzed conversion of aldehydes has been found to be a powerful method.With the continuous development in recent years,it has become an efficient and uncomplicated strategy for constructing ketones.There are four major mechanisms for transition metal-catalyzed ketone synthesis from aldehyde: (1) carbonyl-Heck reaction,that is 1,2-insertion of organometal species to aldehydic C=O double bond,(2) direct insertion of transition metal catalysts to aldehydic C-H bond,(3) aldehyde as acyl radical,(4) aldehyde as carbon radical acceptor.This article summarizes related reports on the transformations of aldehydes to generate corresponding ketones under different reaction conditions.
1.Introduction
Ketones are one of the most important classes of organic compounds,for example,aryl ketones,heteroaryl ketones,and diaryl ketones are common structural motifs in multitudinous natural products [1–9].A series of derivatives of ketones are versatile building blocks for the synthesis of more complex natural products,biologically active molecules,pharmaceuticals,agricultural chemicals,dyes,and functional materials (Fig.1) [10–16].Due to their importance,the development of synthetic methods for the preparation of these scaffolds has received considerable attention.
Traditional methods for the preparation of ketone compounds include Friedel-Crafts reaction of substituted aromatic rings and acylation of organometallic reagents [17–25].However,the above strategies have some disadvantages to a certain extent.These methods are often painstaking,laborious,and time-consuming multistep processes,making the preparations typically inefficient and uneconomical.For example,when using Friedel-Crafts acylation reaction,an excessive amount of Lewis acid catalyst (such as AlCl3) is usually required,and a large amount of acid gas and sewage are generated during the reaction and post-treatment process,causing environmental pollution [19].Moreover,the formation oforthoandparaisomers with untunable regioselectivity results in separation problems and makes aryl ketones withmetasubstituents difficult to access.Consequently,there is a great need to develop simple,effective and versatile methods to synthesize these scaffolds.
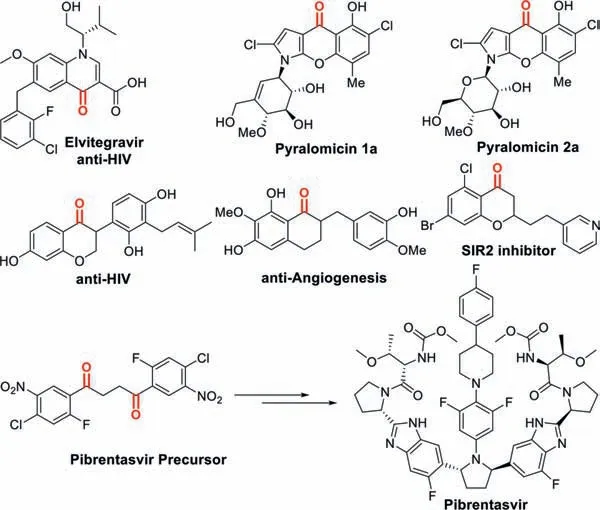
Fig.1.Selective examples of biologically active ketones.
In terms of molecular structure,using aldehyde as starting material to prepare ketone,the conversion would be considered as a much more simple and efficient protocol.And the functionalization of aldehydic C–H bonds has been quite an active research area.However,in the past,the strategy was generally achieved by adding organometallic reagent to aldehyde and then oxidizing the alcohol intermediate (Scheme 1a) [26–30].The operation procedures of this approach are cumbersome and organometallic reagents are sensitive to air,so further developments are needed to realize the generation of ketones from aldehydes through a onestep process.A large number of studies have shown that the synthesis of ketones through the C–H activation and functionalization of aldehydes under the catalysis of various transition-metals is an effective strategy.The direct hydroacylation of olefins and alkynes is a typical reaction of this conversion,which has been summarized by Willis in 2010 [31].Hydroacylation generally involves the addition of an acyl unit and a hydrogen atom across a C–C multiple bond,including both intra- and intermolecular type reactions(Scheme 1b) [31–41].In addition,N-heterocyclic carbene (NHC)catalyzed umpolung additions of aldehydes to carbonyl compounds(benzoin reaction) [42–44],imines (aza-benzoin reaction) [45–49],and Michael acceptors (Stetter reaction) [50–58],are also powerful protocols to produce functional ketones by hydroacylation(Scheme 1c) [59–70].
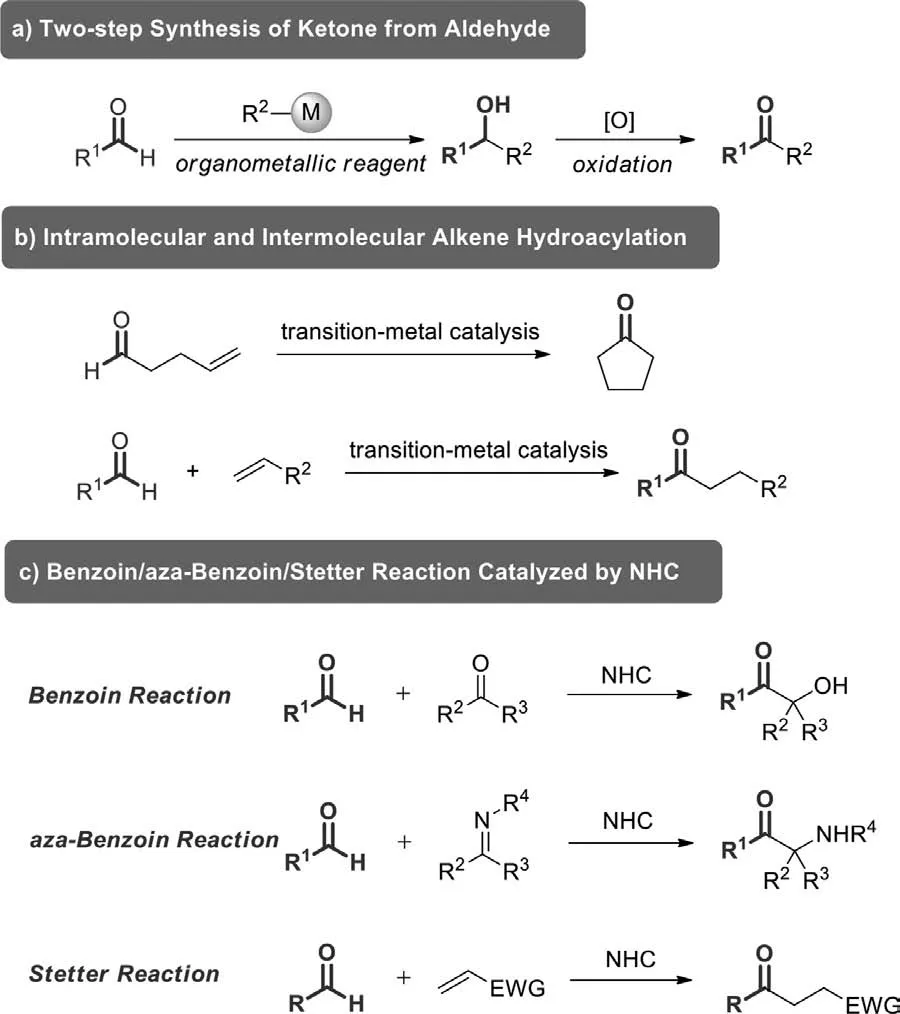
Scheme 1.Selective examples of biologically active ketones and derivatives.
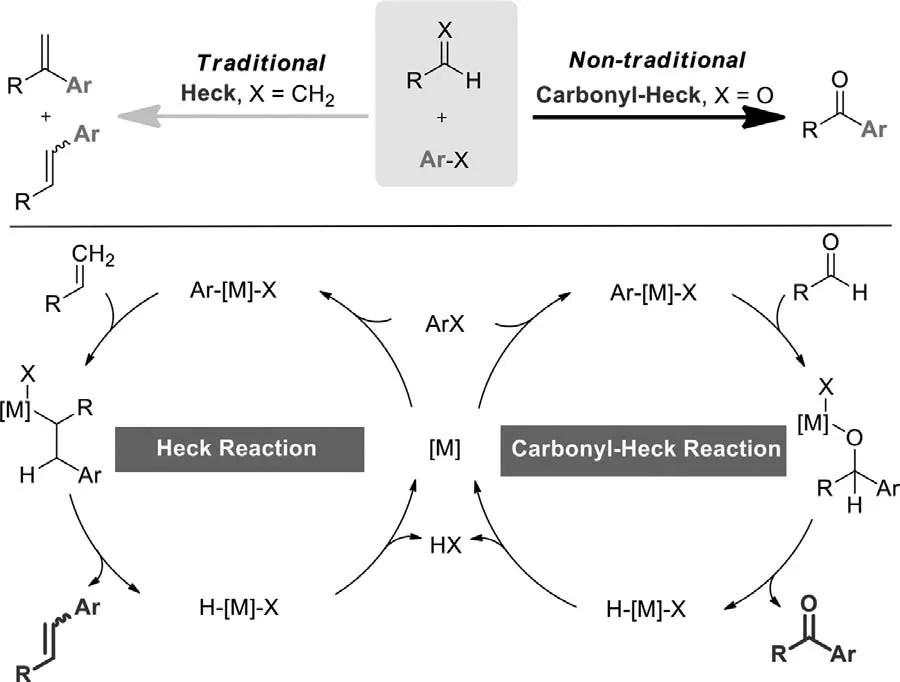
Scheme 2.Heck reaction and carbonyl-Heck reaction.
Over the past few decades,transition metal-catalyzed conversions of aldehydes to ketones have been found to be a powerful method.Despite these significant advances,the development of novel strategies to synthesize various ketone derivatives still drawn considerable attention.With the continuous efforts in recent years,transition metal-catalyzed cross-coupling reactions have been the highly versatile methods for the construction of complex molecules from simple building blocks in both research laboratories and industry.Generally,these reactions are tolerant of a variety of functional groups,applicable to a wide range of substrates,and have excellent practical value and broad application prospects [71–76].In these transformations,the most commonly utilized transition metals may include group VIIB,VII and IB,such as rhodium,ruthenium,palladium,nickel,cobalt,platinum,copper,silver,iron,manganese (Fig.2).Herein,this article summarizes the related conversions using the different catalytic system with transition metals.

Fig.2.Transition metals used in the conversions of aldehydes to ketones.
There are four major mechanisms for transition metal-catalyzed ketone synthesis from aldehyde: (1) carbonyl-Heck reaction,that is 1,2-insertion of organometal species to aldehydic C=O double bond,(2) direct insertion of transition metal catalysts to aldehydic C-H bond,(3) aldehyde as acyl radical,(4) aldehyde as carbon radical acceptor.
2.Carbonyl-Heck-type mechanism
Heck reaction,the coupling of olefins with aryl halides or pseudo-halides,is among the most important transformations in modern synthetic chemistry.In the carbonyl variant of Heck reaction,the C=O bond of aldehyde takes the role of the olefin in the insertion/β-H elimination steps.Conceptually,ketones,instead of substituted olefins,are afforded as the major product of the catalytic cycle (Scheme 2).
2.1.Rh catalysis
In 2000,Hartwig group reported the rhodium(I)-catalyzed coupling reaction of aryl halides with activatedN-pyrazyl aldimines through the Heck-type mechanism (Scheme 3) [77].TheN-pyrazyl aldimines could be easily prepared by the condensation of aldehydes and 2-aminopyrazine,and the ketones were generatedviahydrolysis of the corresponding ketimines.However,the method is not an atom-economic way to access ketones,needing three steps,and theN-pyrazyl aldimines should be prepared in advance.
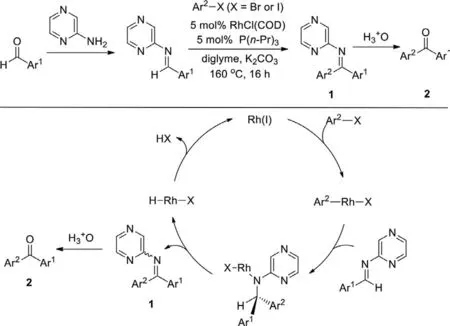
Scheme 3.Synthesis of ketones via the coupling of aryl halides with N-pyrazyl aldimines.
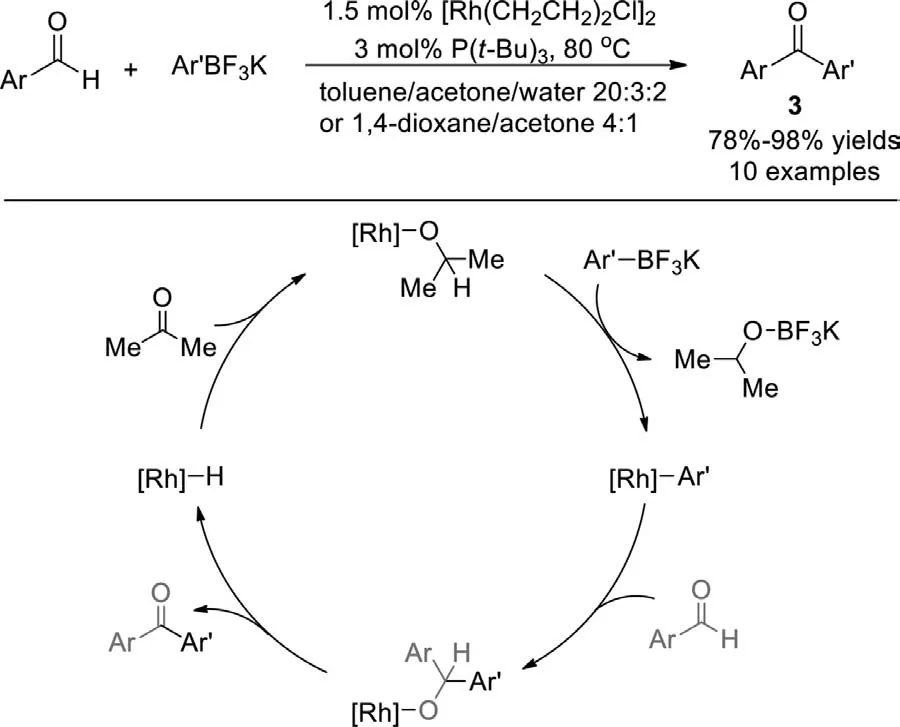
Scheme 4.Rh(I)-catalyzed cross-coupling of arylaldehydes with ArBF3K.
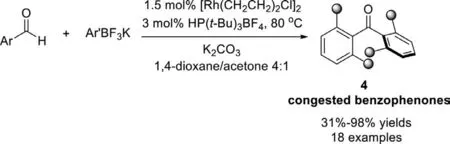
Scheme 5.Sterically hindered benzophenones via Rh(I)-catalyzed cross-coupling of arylaldehydes with ArBF3K.
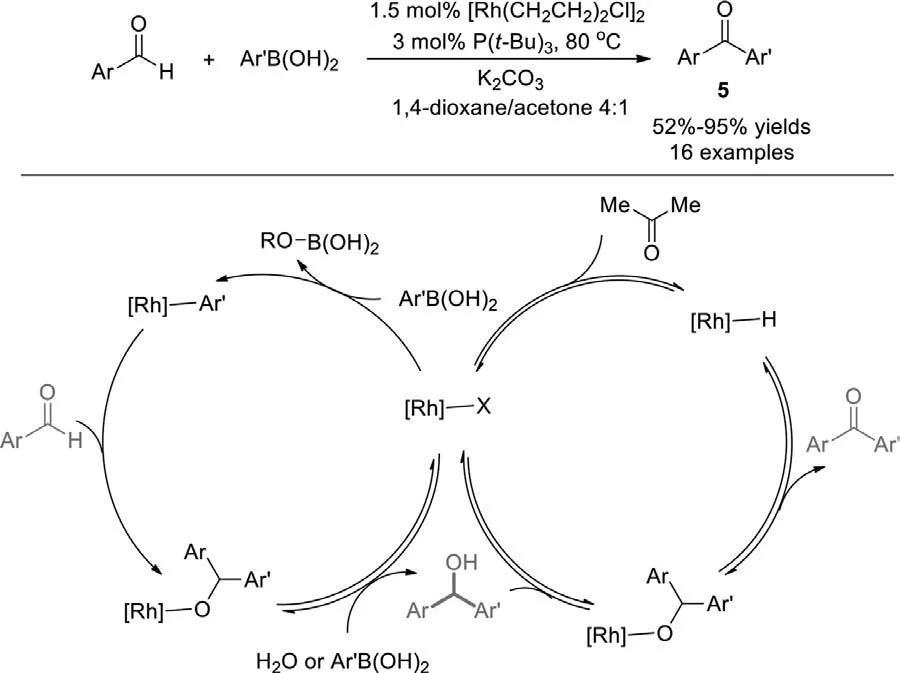
Scheme 6.Rh(I)-catalyzed cross-coupling of arylaldehydes with arylboronic acids.
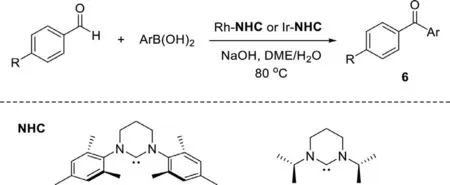
Scheme 7.Arylation of aryl aldehydes with arylboronic acids.
Using P(t-Bu)3as ligand,in 2004,Darses and Genetet al.reported the first rhodium(I)-catalyzed cross-coupling reaction of organoboronic reagents (ArBF3K) with aryl aldehydes to generate diaryl ketones under mild conditions without base (Scheme 4)[78].Deuterium labeling experiment indicated that this transformation proceeded in a Heck-type mechanism,followed by an unusual hydrogen transfer process,in which acetone worked as a hydride acceptor.
However,the majority of biologically active benzophenones are sterically congested,at least bearing di-ortho-substituents,and were mainly obtainedviathe addition of aryllithium or arylmagnesium to aldehydes followed by oxidation.The highly desirable mild transition-metal-catalyzed reactions are limited to reagents that are sterically unencumbered.In 2008,Darses and Genet group employed a rhodium/tri-tert-butylphosphane catalyst system with the stable phosphonium salt as the precursor of P(t-Bu)3instead of highly oxidizable P(t-Bu)3,and for the first time disclosed a straightforward protocol for the synthesis of congested benzophenones from aryl aldehydes and potassium aryltrifluoroborates (Scheme 5) [79].This reaction was handled in easy-to-operate conditions.A broad substrate scope was tolerated,di-,tri-,and even tetra-ortho-substituted benzophenones can be synthesized.
In 2007,Genetet al.employed arylboronic acids and developed the rhodium(I)-catalyzed cross-coupling with aryl aldehydes to access directly ketonesviaformal C-H bond activation(Scheme 6) [80].The reaction pathway was different from their above-mentioned work using ArBF3K which was a tandem catalytic process involving two cycles,an addition cycle and an oxidation cycle combined throughβ-hydride transfer.In GC/MS monitoring experiment,surprisingly,the aldehyde disappeared very fast to form carbinolviathe protonation of alkoxo-rhodium intermediate by water formed in the reaction medium (or directly by the boronic acid),giving an active hydroxo-rhodium species.It demonstrated that this reaction may include two consecutive catalytic reactions: rhodium-catalyzed addition of the boronic acid to aldehyde to produce the carbinol;oxidation of the corresponding alcohol to provide the ketone.
In 2004,Buchmeiser and co-workers reported Rh- or Ir-NHC complexes catalyzed arylation of aryl aldehydes with arylboronic acids which described a straightforward access to diaryl ketones with low catalyst loading (0.08-1 mol%) (Scheme 7) [81].The structure of catalyst strongly affected the activity and selectivity of the reaction,the tetrahydropyrimidin-2-ylidene ligands enhance the efficiency;the propyl groups at both nitrogen atoms of NHC ligands are more effective than other substituents;iridium shows low activity but high selectivity.
In 2019,Krische group developed a regiodivergent rhodiumcatalyzed reductive coupling-redox isomerization of aldehydes with vinyl bromide to form branched (PtBu2Me) or linear (PPh3)ketones using different ligands (Scheme 8) [82].A series of deuterium labeling experiments disclosed that a redox isomerization was the critical step.Initially,the oxidative addition of vinyl bromide formed vinyl rhodium(III) species.In the process of the formation of linear ketone,the weakσ-donor ligand PPh3dissociated at the vinylrhodium(III) intermediate stage and motivated to form transient allene throughβ-hydride elimination,and then generated allylrhodium(III) hydride species.Aldehyde insertion provided a homoallylic rhodium alkoxide,and the bromide moiety of the kinetic rhodium alkoxide reacted with HCO2K to form a hydride.Followed by reductive elimination to afford a homoallylic alcohol which went through isomerization to deliver the linear ketone.For the formation of branched ketone,the strongσ-donor ligand was used.The aldehyde inserted to vinyl rhodium(III) species,converted to the allylic alcohol and,therefrom,the branched ketone in the analogous process.
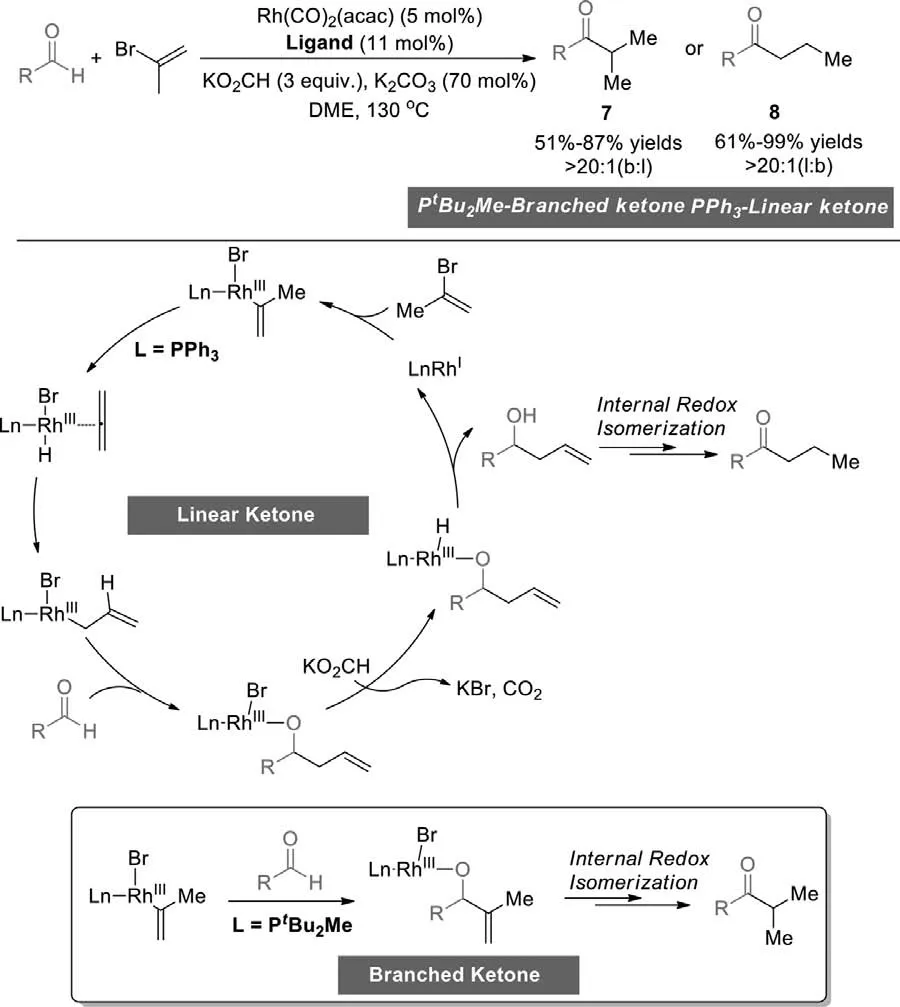
Scheme 8.Regiodivergent Rh-catalyzed vinyl bromide reductive coupling-redox isomerization.
Later,employing the same catalyst system for the branched ketones,and replacing vinyl bromide with vinyl triflate,they developed the vinyl triflate-aldehyde reductive coupling-redox isomerization (Scheme 9) [83].Many functional aryl or alkyl aldehydes afford the corresponding ketones under the conditions as the previous vinyl bromide adopted.
2.2.Pd catalysis
In 2008,Wu and Chenget al.also reported the synthesis of diaryl ketones using a palladium-catalyzed coupling of aromatic aldehydes with aryl boronic acids in moderate to excellent yields(Scheme 10) [84].The reaction proceeded smoothly in the presence of Pd2(dba)3and P(1-nap)3.It is worth noting that Cs2CO3might exhibit a dual ability: (1) Facilitating the addition of organoboronic acids to aldehydes,affording carbinol derivatives;(2) Prompting the aerobic oxidation of the carbinol derivativesin situto aromatic ketones.A variety of functional groups like nitro,cyano,acetoxy,heteroaryl and steric substrates were all well tolerated to access to the desired ketones in good efficiency.All the reactions were carried out under an ambient atmosphere,and rigorous exclusion of air or moisture was not required.
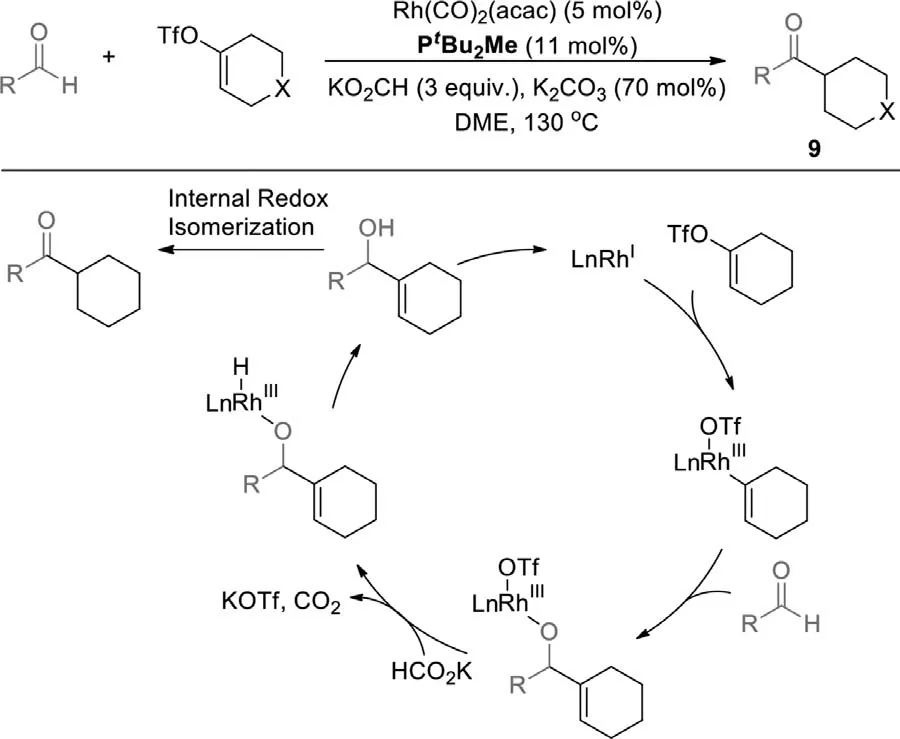
Scheme 9.Rh-catalyzed vinyl triflate-aldehyde reductive coupling–redox isomerization.
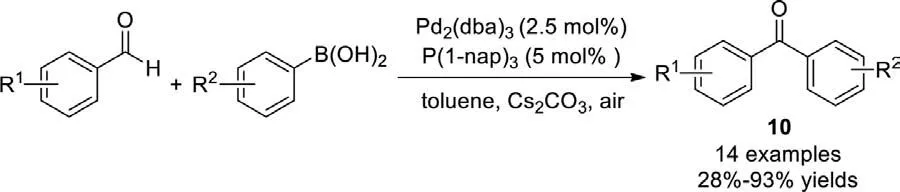
Scheme 10.Pd-catalyzed coupling of aldehydes and organoboronic acid.
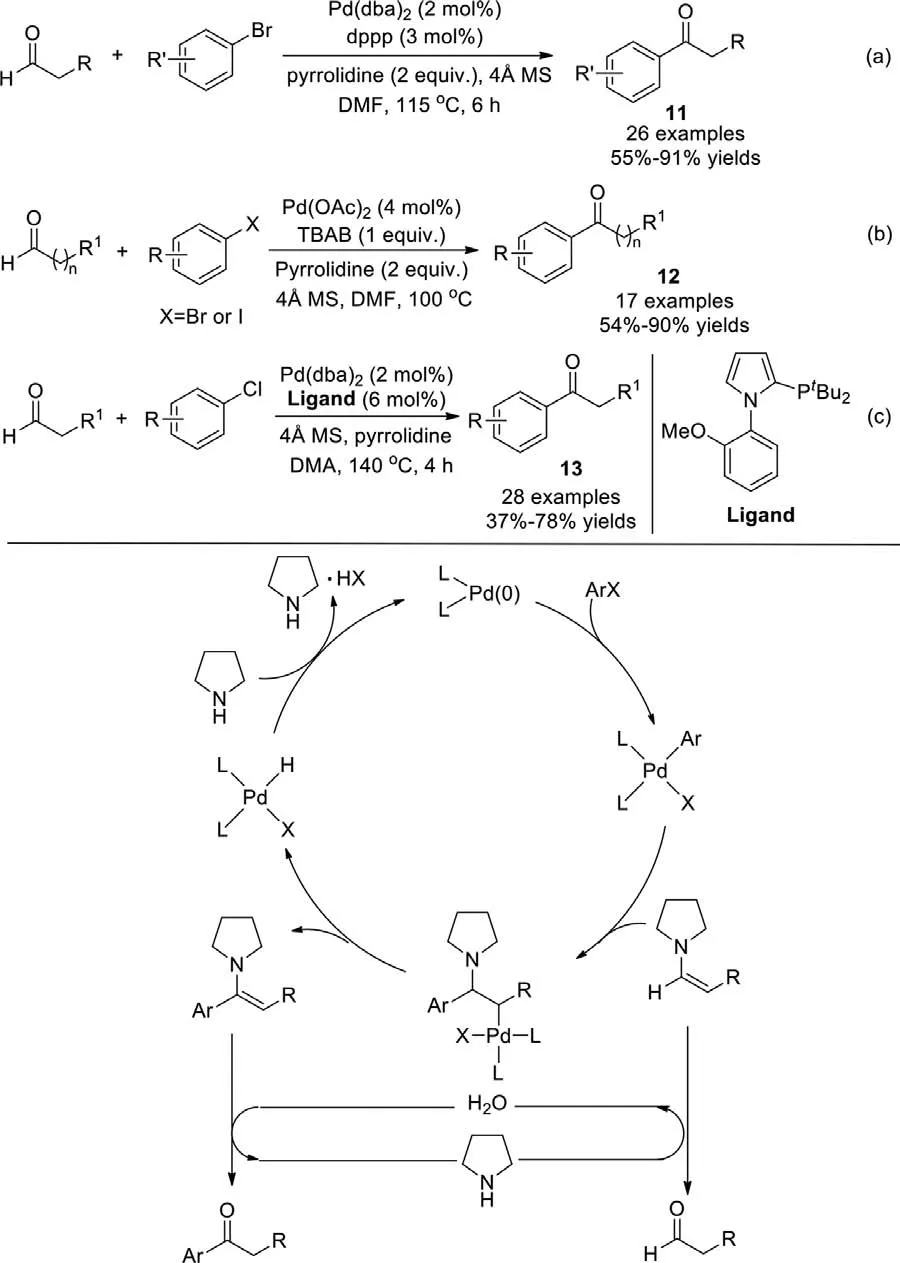
Scheme 11.Pd-catalyzed direct acylation of aryl halides.
In 2008,a direct acylation of aryl bromides was developed by Xiao and co-workers,affording alkyl aryl ketones in one step,using palladium and pyrrolidine as the co-catalysts and 4 ˚A MS as additive (Scheme 11a) [85].In the presence of alkyl aldehydes,the addition of pyrrolidine resulted in the formation of a highly reactive electron-rich enamine,subsequently a Heck-type reaction with palladium,followed by hydrolysis to form the corresponding ketones.It is interesting to note that,under similar conditions but without pyrrolidine and 4 ˚A MS,the coupling of aldehydes with aryl bromides led toα-arylated aldehydes instead of ketones.
Scheme 12.Heteroaryl and diheteroaryl ketones synthesis via Pd-catalyzed 1,2-addition and oxidation.
Furthermore,a palladium(0) nanoparticle-catalyzed direct acylation aryl bromides or iodides was reported by Ranu group in 2010 [86].Tetrabutylammonium bromide (TBAB) was used as an additive,which stabilized the Pd nanoparticle from gathering.Various aryl iodides and bromides proceeded smoothly with openchain aldehydes to form the alkyl aryl ketones in good yield(Scheme 11b).A few months later,Xiao group extended the substrate to a wide range of aryl chlorides in high temperature,a bulky electron-rich monophosphine ligand was adopted to enhance the reaction activity,and the reaction was tolerant of functionalities on the aliphatic aldehydes (Scheme 11c) [87].
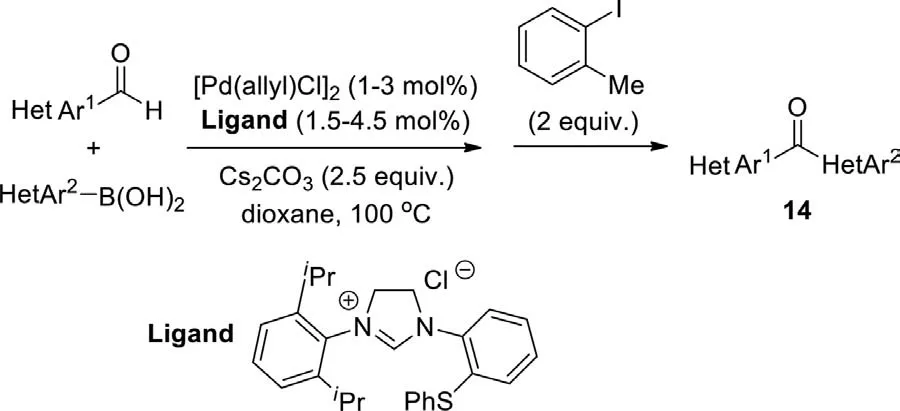
In 2013,Kuriyama and Onomuraet al.developed the palladium/imidazolinium carbene-catalyzed 1,2-addition and oxidation to synthesize heteroaryl and diheteroaryl ketones from aldehydes and organoboronic acids (Scheme 12) [88].The 1,2-addition involves a transmetalation-insertion mechanism,and the oxidation proceeds through oxidative addition,β-hydrogen elimination,and reductive elimination.A high level of catalyst performance was attained in the one-pot process using 1.0-3.0 mol% catalyst loading.Several oxidants were examined,a sterically hindered 2-iodotoluene was the optimal choice,giving the desired ketones in good to excellent yields.
2.3.Ni catalysis
Although Rh-,Ru- and Pd-catalyzed coupling reactions for ketones synthesis from aldehydes were well established,efforts have been made to replace these expensive metals.Owing to its high activity showed in many coupling reactions,nickel is considered as the most promising cheap metal.The first nickel-catalyzed crosscoupling reaction of aldehydes to access ketones was reported by the Cheng group in 2002,providing a new way for chemoselective synthesis of diaryl ketones using aryl aldehydes and aryl iodides in fair to good yields (Scheme 13) [89].The catalytic reaction proceeds smoothly only with bidentate phosphine nickel complexes as catalysts,such as Ni(dppe)Br2,whereas monodentate phosphine complexes are completely ineffective.

Scheme 13.Ni-catalyzed ketone synthesis via coupling of aldehydes and aryl iodides.
Scheme 14.Ni-catalyzed C-H arylation of 2-hydroxybenzaldehydes with aryl iodides.
It is worth noting that zinc powder is critical to the coupling.Reduction of Ni(II) to Ni(0) by zinc metal with the formation of zinc halide likely initiates the catalytic reaction.The oxidative addition of aryl iodide,coordination of aldehyde,formation of nickel alkoxide and subsequentβ-hydride elimination occurs,and affords the desired ketone.
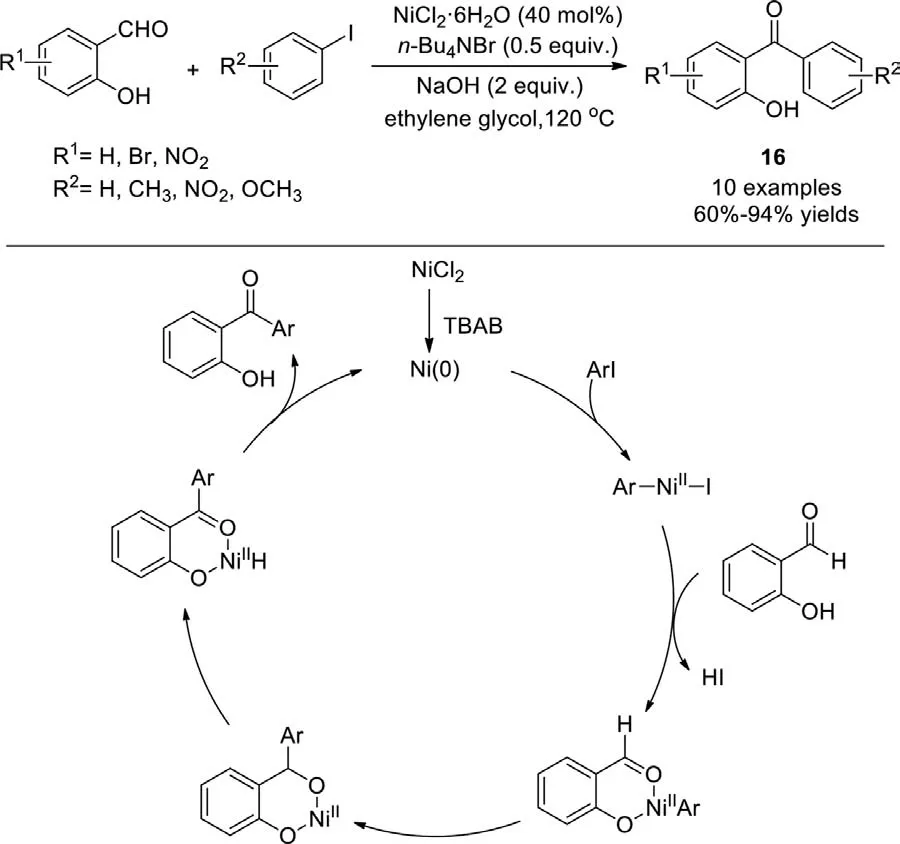
In 2015,the ligand-free nickel-catalyzed C-H arylation of 2-hydroxybenzaldehydes with aryl iodides to access 2-hydroxybenzophenones was reported by Nowrouzi group,employing the catalyst system of NiCl2·6H2O andn-Bu4NBr (Scheme 14)[90].Although only 10 examples,this is an efficient,inexpensive,and practicable method for the synthesis of ketones.It is worth noting that the introduction ofn-Bu4NBr as an additive not only stabilizes the catalytic system by avoiding undesired agglomeration and deactivationviathe formation of a monomolecular layer around the metal core,but also plays as a reductant for the generation of Ni(0).
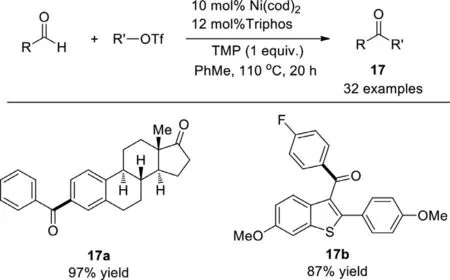
In 2017,Newman and co-workers developed the nickelcatalyzed carbonyl-Heck reaction of aldehydes with organotriflates to make ketones,using Ni(0) precatalyst,tridentate triphos ligand,and bulky amine base (TMP,2,2,6,6-tetramethylpiperidine)(Scheme 15) [91].This pathway is supported by the effectiveness of the catalyst in traditional Heck reactions.High-throughput experiment was utilized to uncover the specific combination of halide/pseudohalide,metal,ligand,and base necessary to enable the reaction.The intermolecular coupling demonstrated extensive compatibility,both aryl aldehydes and aryl triflates bearing electron-donating,electron-withdrawing,and sterically hindering functionality were tolerated.Aliphatic aldehydes need quinuclidine as the base to prevent Tishchenko-type side products when using TMP.Some bioactive molecules can be prepared,such as estrone-derived compound 17a and benzothiophene 17b (precursor to Raloxifene).
Scheme 15.Ni-catalyzed carbonyl-Heck reaction of aldehydes with organotriflates.
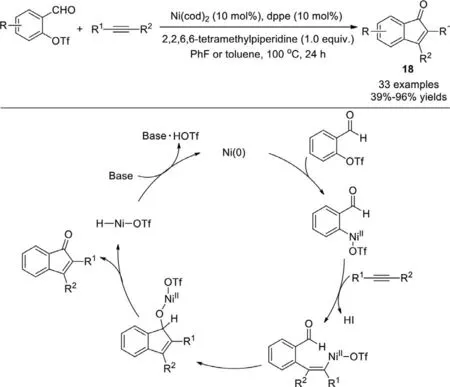
Scheme 16.Ni-catalyzed Larock annulations of substituted 2-formylphenyl trifluoromethanesulfonate with alkynes.
Recently,we present a direct transformation based on nickelcatalyzed Larock annulations of substituted 2-formylphenyl trifluoromethanesulfonate with alkynes to deliver a wide range of indenone analogs that are valuable synthetic intermediates for natural compounds (Scheme 16) [92].This catalytic system is shown to proceed through a redox neutral arylation in high yields with excellent regioselectivities.Mechanistically,the oxidative addition of a nickel into the weak C–OTf bond,followed by the migratory insertion of the alkyne,and subsequently the intramolecular “carbonyl” variant of the Heck reaction andβ-H elimination leads to the formation of the desired indanone.
2.4.Ru catalysis
The first Ru-catalyzed cross-coupling reaction of aldehydes with arylboronic acids to generate aryl ketones was described by Wan group in 2011,aliphatic and aromatic aldehydes are effective(Scheme 17) [93].The kinetic isotope effect experiment demonstrated that C–H activation of aldehyde was not involved in the catalytic cycle,and hydrogen abstraction from alcohol was the probably rate-limiting step.The possible reaction pathway was initiated by transmetalation of boronic acid with [Ru(CO)3Cl2]2to give aryl ruthenium complex,then aldehyde inserted into ruthenium-carbon bond to provide an alkoxo-ruthenium intermediate 20 which underwentβ-hydride elimination to afford the final ketone and ruthenium hydride complex.Subsequently,pinacolone inserted into the ruthenium-hydrogen bond to generate alkoxo-ruthenium intermediate 21,followed by transmetalation wtih boronic acid to regenerate arylruthenium complex.
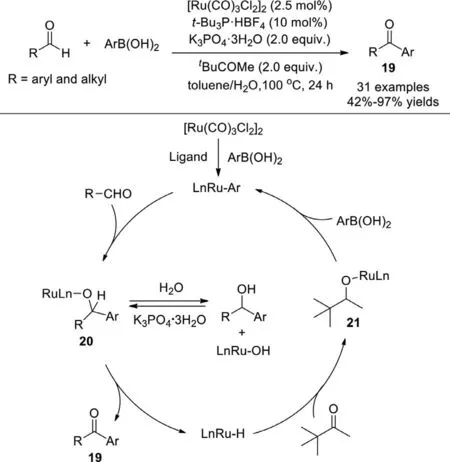
Scheme 17.Ru-catalyzed cross-coupling of aldehydes with arylboronic acids to generate aryl ketones.
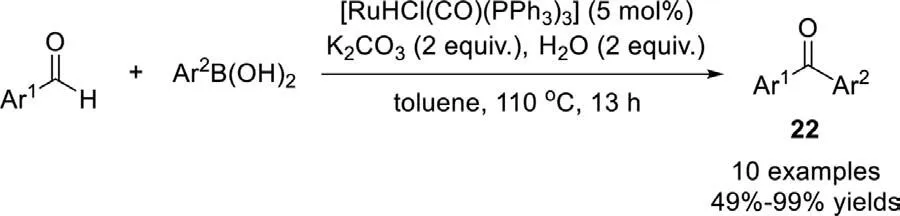
Scheme 18.Ru-catalyzed coupling reaction of arylboronic acids with aryl aldehyde.
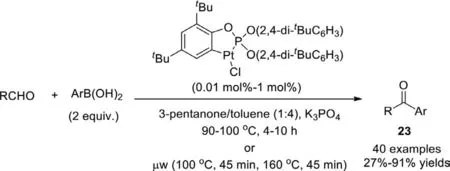
Scheme 19.Orthoplatinated triarylphosphite-catalyzed ketone synthesis.
In the same year,Fukuyama and Ryuet al.also reported a ruthenium-catalyzed coupling reaction of aryl aldehydes with arylboronic acids for the synthesis of diaryl ketones using[RuHCl(CO)(PPh3)3]as catalyst (Scheme 18) [94].Various arylboronic acids and aryl aldehydes especially the steric 2-substituted aryl aldehyde work smoothly to give the desired ketones in good yields.This reaction underwent a hydrogen transfer pathway and may contain two consecutive steps: formation of the alcohols and hydrogen transfer to aldehydes to form ketones.Without water,the reaction did not proceed,and no ketone was formed.

Scheme 20.Co or Cu-catalyzed arylation of arylboronic acids with aldehydes.
2.5.Pt catalysis
In 2010,Hu and co-workers reported an addition reaction followed by an oxidation of aldehydes with arylboronic acids to access aryl ketones (Scheme 19) [95].The reaction employed orthoplatinated triarylphosphite as catalyst and K3PO4as the best base.The catalyst is an anionic four-electron donor-based metallacycle,which is readily available and air/moisture-stable.3-Pentanone was identified as a suitable oxidant for the oxidation of alcohol step.Notably,aryl and aliphatic aldehydes can react well with the arylboric acid to obtain the corresponding aryl-aryl and alkyl-aryl ketones in good yield.Besides,a microwave-assisted method was developed with low catalyst loading (0.01 mol%),and the reaction time shortened dramatically.The platinacycle-catalyzed protocols represent the first examples of direct access to aryl alkyl ketones from alkyl aldehydes with arylboronic acidsviathe tandem addition-oxidation reaction.
2.6.Co and Cu catalysis
A cobalt-catalyzed arylation of arylboronic acids with aldehydes was developed by the Cheng group in 2011,in the presence of CoCl2and tmphen under 1 atm of O2(Scheme 20a) [96].Both electron-withdrawing and electron-donating substituted aryl aldehydes and heterocyclic aldehydes reacted smoothly with different substituted boronic acids to afford diaryl ketones in good yields.This cobalt-catalyzed reaction may proceed in a Heck-type mechanism,a cobalt hydride was formed,O2as an oxidant to regenerate the active Co(II) species.
Chen and Wuet al.achieved this tramsformation using the Cu(OTf)2and Xantphos catalytic system,aryl ketones were formed in moderate to good yields (Scheme 20b) [97].As oxidant,oxygen was critical for the reaction.Different aryl aldehydes were well tolerated irrespective of the electronic effect of substituents.However,electron-withdrawing arylboronic acids reacted slowly in lower yields than electron-rich ones owing to the weak transmetalation ability.Although this reaction showed limited examples,the cheap copper was used and a practicable way for the synthesis of ketones was established.
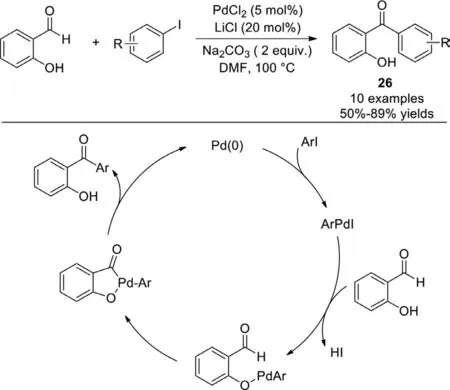
Scheme 21.Pd-catalyzed coupling of 2-hydroxybenzaldehyde with aryl iodide.
Scheme 22.Pd-catalyzed coupling of 2-hydroxybenzaldehyde with aryl boronic acids or aryl halides.
3.Insertion of metal catalysts to aldehydic C-H bonds
In combination with these reports on the transition-metal catalyzed carbonylative Heck reaction of aryl halides,pseudohalides or organometallic reagents,this methodologyviadirect insertion of metal catalysts to aldehydic C−H bond affords another way to complete the Heck reaction on carbonyl groups.Most importantly,it provides efficient synthetic method for preparing the structural motifs of valuable natural products and bioactive molecules such as aurone,flavone,2′-hydroxychalcone,and flavanone.
3.1.Pd catalysis
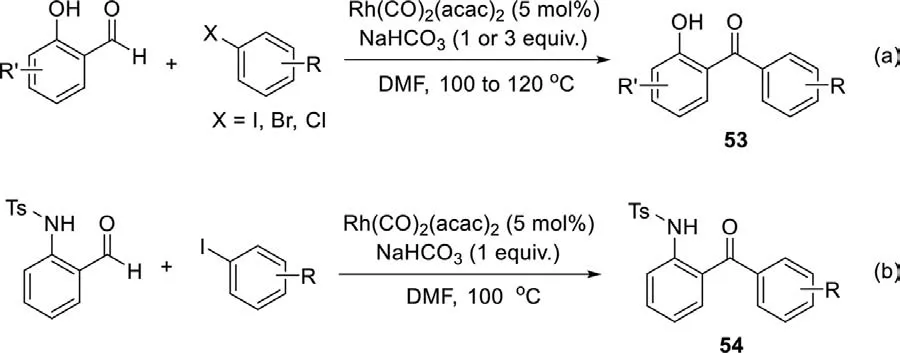
As early as 1996,Miura and co-workers reported the first palladium-catalyzed coupling of 2-hydroxybenzaldehydes with aryl iodidesviachelation-assisted C−H cleavage of the aldehyde [98].The direct PdCl2-catalyzed ketone synthesis from aldehyde proceeded smoothly in the presence of Na2CO3(base) and LiCl(additive),although the scope was limited in several examples (Scheme 21).In the plausible catalytic cycle,oxidative addition of aryl iodide to Pd(0) to produce the aryl palladium species which reacted with 2-hydroxybenzaldehyde to generate the aryl(aryloxy)palladium intermediate and released HI.Then,oxidative addition produced the palladium(IV) species,followed by twofold reductive elimination to afford the corresponding ketone.
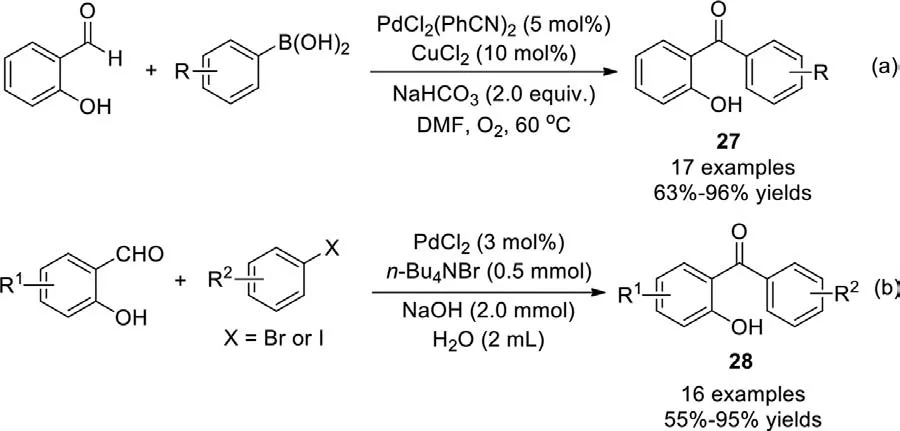
A similar strategy using arylboronic acid was developed by Xu group in 2010,and various functional 2-hydroxybenzophenones were synthesized under mild conditions in good to excellent yield(Scheme 22a) [99].This reaction required the use of O2as an oxidant to convert the alcohol intermediate into the corresponding ketone.In 2015,aryl iodides,aryl bromides and benzyl bromides were applied in the synthesis of ketones in neat water by Nowrouzi group,no oxidant was needed (Scheme 22b) [100].
In 2005,Chang and co-workers reported the Pd and Ru cooperative catalysis to synthesize ketonesviathe direct coupling of 8-quinolinecarboxaldehyde with iodoarenes or organostannanes[101].The C-H bond of the aldehyde was activated by ruthenium to generate five-membered cycloacylruthenium hydride intermediate,and then reacted with the aryl palladium species,followed by reductive elimination,the desired ketone was formed finally.The reaction exhibited wide functional group compatibility to aryl iodides and stannanes,ketones were afforded in moderate to good yields (Scheme 23).The transformation demonstrated that the appropriate combination of two compatible metallic catalysts provides a new possibility for the development of novel synthetic methodologies that are difficult to carry out with any singlecatalyst systems.

Scheme 23.Pd and Ru co-catalyzed ketone synthesis from aldehyde with iodoarenes or organostannanes.

Scheme 24.Pd-catalyzed cross-coupling of N-tert-butylhydrazones with aryl bromides.
In 2006,Hartwig group developed palladium-catalyzed arylation ofN-tert-butylhydrazones with aryl bromides,employing Pd2(dba)3and DPEphos as catalyst [102].These hydrazones,served as acyl anion equivalents,are readily accessible from aldehydes.Under mild conditions,theN-tert-butylhydrazones could undergo cross-coupling at the C-position of the diazaallyl unit,isomerization of the initial azo product to the corresponding hydrazones,followed by hydrolysis,would generate the arylketones (Scheme 24).
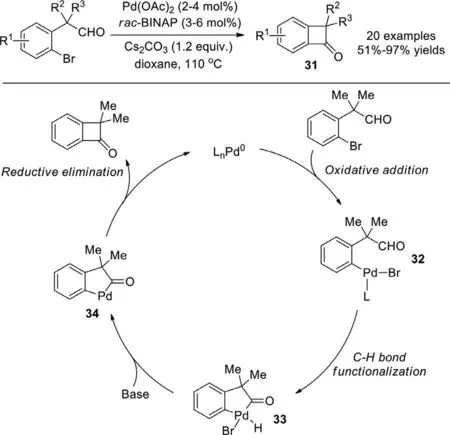
Owing to the inherent ring stain,active benzocyclobutenone(BCB),a powerful synthetic intermediate,was widely used for many transformations like cycloaddition,C−C bond activation.Previous preparations of this BCB were achieved mainly through[2+2]cycloadditions,metal-mediated intramolecular cyclizations,however,functional BCBs cannot easily be obtained from these traditional strategies.In 2010,Martin and co-workers reported a more practicable way for the synthesis of BCBs with intramolecular acylation of aryl bromidesviaC−H functionalization (Scheme 25)[103].Various substrates with different substituents like alkene,ester,nitrile,aldehyde,ketone,free hydroxy,silyl groups,alkyl halide can be accommodated in this reaction.In the plausible pathway,oxidative addition of Pd(0) to C−Br bond formed intermediate 32,followed by C−H functionalization to afford intermediate 33 and then released the HBr to generate a five-membered metallacycle 34 under base,finally the reductive elimination gave the corresponding benzocyclobutenone 31.In addition,the kinetic isotope effect experiment demonstrated that C−H bond cleavage was a rate-limiting step.
Scheme 25.Pd-catalyzed intramolecular acylation of aryl bromides to synthesize benzocyclobutenones.
Scheme 26.Pd-catalyzed C−H activation of aldehydes and (hetero)aryl halides to synthesize diaryl ketones.
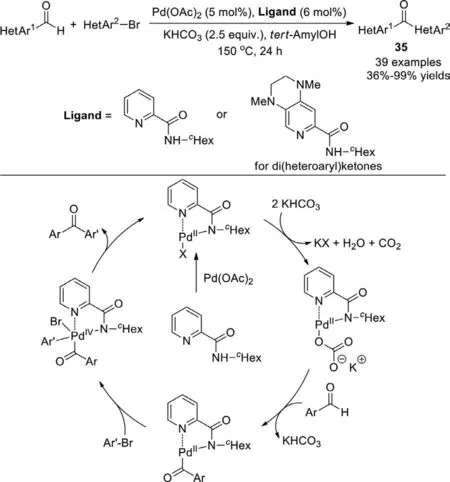
In 2018,the Kuninobu and Kanaiet al.developed a practical procedure for the synthesis of diaryl ketones from aldehydes and (hetero)aryl halidesviapalladium-catalyzed C−H activation (Scheme 26) [104].The picolinamide ligand is the key for this reaction,which activates the C−H bond of aldehyde through a palladium-amide intermediate,then oxidative addition of aryl bromides to this intermediate,eventually a reductive elimination to form the desired ketone.With respect to the substrates,various aryl bromides with aryl or heteroaryl aldehydes were tolerated,and desired diaryl ketones or di(heteroaryl) ketones were formed in good yields,even in gram-scale.The transformation also proceeded using aliphatic aldehydes,whereas the yields of the corresponding ketones were low.Besides,the reaction was applied to the synthesis of Fenofibrate,a commercial drug used to reduce cholesterol levels in patients at risk for cardiovascular disease.
Scheme 27.Arylation of α,β-unsaturated aldehydes with arylboronic acids.
3.2.Rh catalysis
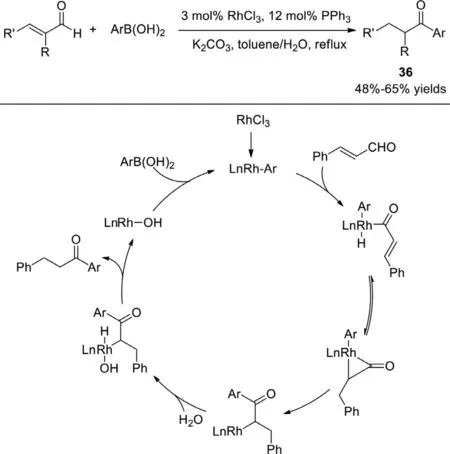
Besides the reported palladium catalysts,rhodium is likewise regularly utilized to catalyze the synthesis of ketones from aldehydes,and many research groups have reported on this.In 2004,an unanticipated self-conjugate reduction and cross-coupling tandem reaction ofα,β-unsaturated arylaldehydes with arylboronic acids was reported by Zou Group,producingβ-phenylpropiophenones as the desired products (Scheme 27) [105].The pathway catalyzed by the RhCl3/PPh3system possibly contains an oxidative addition of the aldehydic C–H bond ofα,β- unsaturated aldehyde to the Rh–Ar species that is generatedin situby reduction of RhCl3with arylboronic acids,followed by a three-membered metallocyclopropanone intermediate,subsequently a reductive elimination,oxidative addition of water,and another reductive elimination to afford the desired product.
In 2008,Satoh and Miuraet al.reported the direct oxidative coupling of salicylaldehydes with internal alkynes to produce 2,3-disubstituted chromone derivatives (Scheme 28) [106].A rhodium catalyst in combination with a cyclopentadiene ligand and a copper oxidant promote this straightforward annulation.During the reaction process,the coordination of the phenolic oxygen atom to an RhIIIX3species gives a rhodium(III) phenolate.Directed C−H rhodation to form a rhodacycle intermediate is then followed by alkyne insertion and reductive elimination to form the desired chromone.The aldehydic C−H bond-cleavage step may be promoted effectively by the metal-directing hydroxy oxygen atom.
In 2011,Li group disclosed the rearrangement of 2-aryloxybenzaldehydes to 2-hydroxybenzophenones by rhodiumcatalyzed cleavage of aryloxy C−O bonds (Scheme 29) [107].This reaction can tolerate a variety of functional groups,thereby indicating the wide potential of applications.Initially,the aldehydic C−H bond insertion by RhIgenerates the RhIIIhydride species 39.Upon reaction withtert-butyl peroxide (TBP),the RhIIIcomplex 40 is formed,thus liberatingtBuOH.Then complex 40 may undergo an intramolecular SNAr process to afford the intermediate 41,thereby generating the RhIIIcomplex 42,which could release acetone and RhIII/Me complex 43.Finally,the reaction of the previously liberatedtBuOH with complex 43 can afford the desired product 38 and form [RhIII(Me)-(tOBu)],which could regenerate the RhIcatalystviaa reductive elimination and releasetBuOMe.
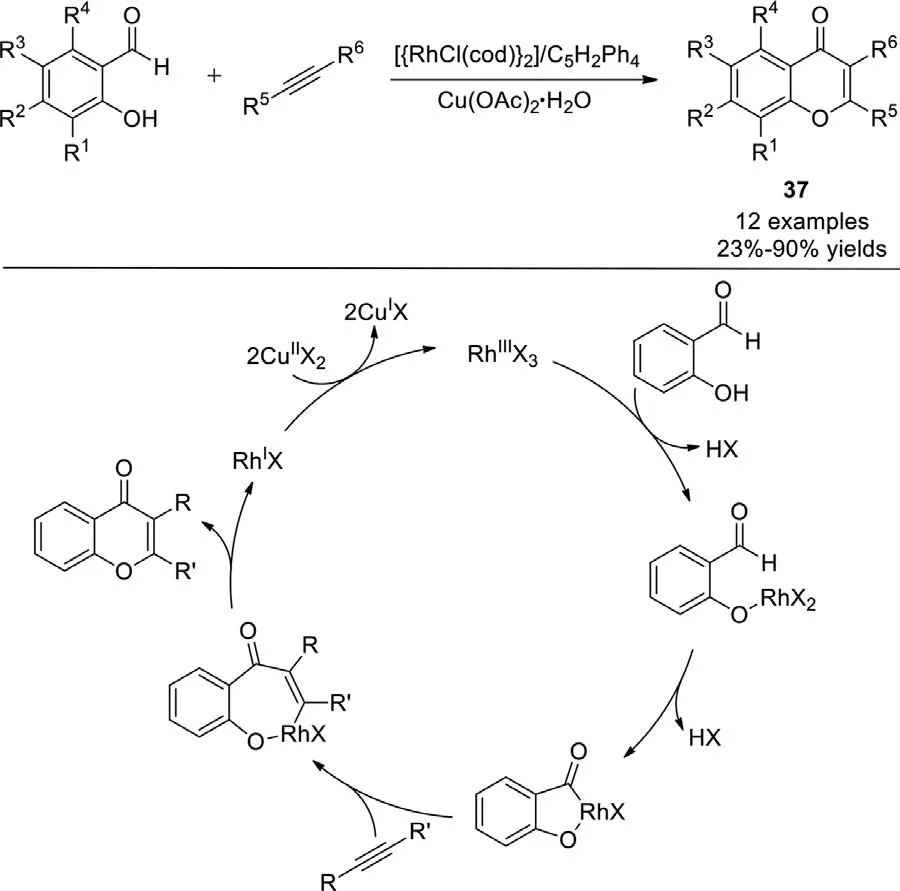
Scheme 28.Rh-catalyzed oxidative coupling of salicylaldehydes with internal alkynes.
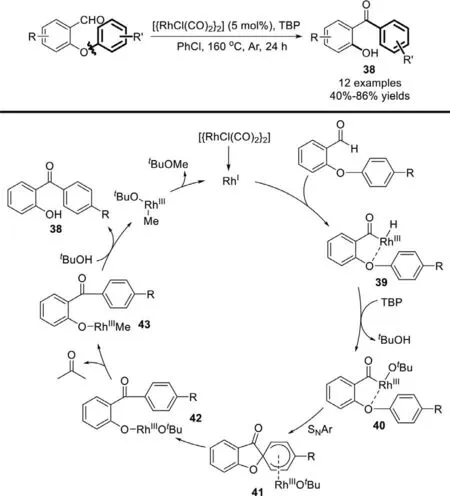
Scheme 29.Rh-catalyzed rearrangement of 2-(aryloxy)-benzaldehydes to 2-hydroxybenzophenones.
In 2012,Glorius and co-workers developed rhodium(III)-catalyzed regioselective dehydrogenative Heck-reaction (DHR) of salicylaldehydes with different classes of olefins (Scheme 30) [108].A mechanism was proposed in Scheme 31.In the presence of the catalyst [(Cp∗RhCl2)2],the products resulted from DHR of aldehydic C−H bonds and subsequent intramolecular oxidative cyclization of C−O bonds.This methodology has been applied to the synthesis of lonchocarpin (45) and 4-methoxylonchocarpin (46) in just two steps from commercially available reagents.They exhibit a range of biological and pharmacological properties including antimutagenic,antimicrobial,antiulcer,and antitumor activities.
In the presence of an Rh/Cu catalyst system,Pihko and Radhakrishnanet al.developed an efficient one pot strategy for the synthesis of cyclopentene fused chromanone derivativesviathe direct oxidative coupling of salicylaldehydes with azabicyclic olefins in 2013(Scheme 31) [109].This is the first report on the ring-opening/ringclosing of azabicyclic olefins through a transition-metal catalyzed protocol involving aldehyde C−H cleavage andπ-allyl chemistry.
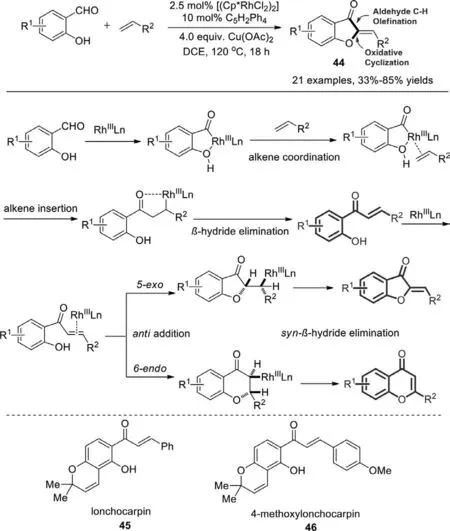
Scheme 30.Rh(III)-catalyzed cyclization of salicylaldehydes and electron-deficient olefins.
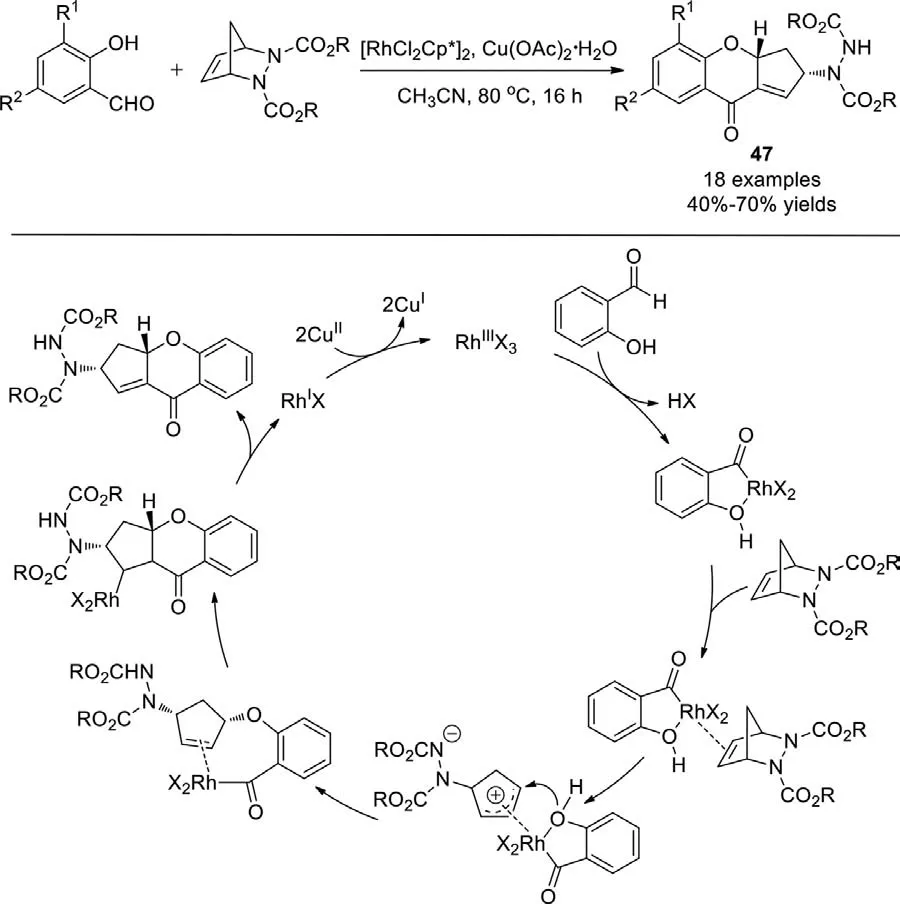
Scheme 31.Oxidative coupling of salicylaldehydes with azabicyclic olefins.
In 2015,Cui group reported Rh(III)-catalyzed salicylaldehydes C−H bond functionalization with arylboronic acids,with features of mild reaction condition and high efficiency (Scheme 32) [110].Furthermore,the functionalized 2-hydroxybenzophenone could be subject to divergent synthesis of heterocycles.
In 2015,Li group achieved Ir(III) or Rh(III)-catalyzed synthesis of ynonesviaC−H activation of benzaldehydes in the coupling with hypervalent iodine alkynylating reagents (Scheme 33)[111].Rhodium and iridium catalysis exhibited complementary substrate scope.The iridium(III) catalyst offered high activity for salicylaldehyde substrates,while the rhodium(III) catalyst is necessary when the substrates wereN-sulfonyl-2-aminobenzaldehydes.In these catalytic systems,the electrophilic aldehyde group is converted to a nucleophilic metal−formyl group,which matches the electrophilic EBX reagents.
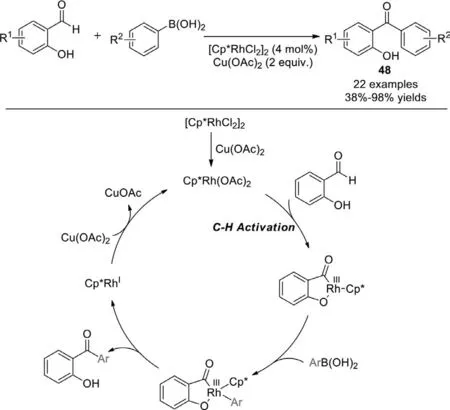
Scheme 32.Rh(III)-catalyzed aldehyde functionalization of salicylaldehydes with arylboronic acids.
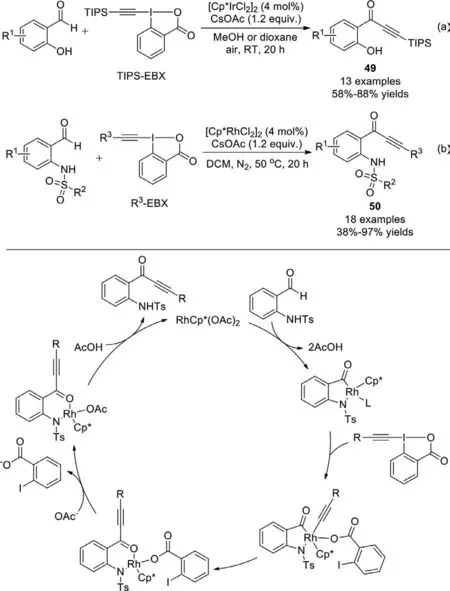
Scheme 33.C−H alkynylation of salicylaldehyde and N-sulfonyl-2-aminobenzaldehyde.
Using the similar catalyst systems,Waser group developed the C−H heteroarylation of salicylaldehydes andN-sulfonyl-2-aminobenzaldehydes with benziodoxolone hypervalent iodine reagents in 2018 (Scheme 34) [112].The resulting salicyloyl indoles and (2-sulfonamino)benzoyl indoles were easy to be transformed into other useful building blocks.
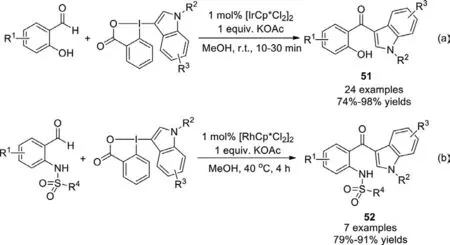
Scheme 34.C−H heteroarylation with benziodoxolone hypervalent iodine reagents.
Scheme 35.Rh-catalyzed directing-group-assisted C–H arylation of aldehydes.
Employing the similar substrates,Rao group reported the directing-group-assisted C−H arylation of aldehydes in 2017,to synthesize medicinally valuable 2-hydroxybenzophenones or 2-(ptoluenesulfonylamido)benzophenones (Scheme 35) [113].Multiple functional aldehydes and bulky aryl halides were suitable for the reaction conditions,producing the corresponding ketones in high efficiency.This transformation was initiated by oxidative addition of aryl halides to form arylrhodium species,which sequentially coordinated to the directing atom of aldehydes.Then,C−H activation and reductive elimination occurred,affording benzophenones.
As reviewed above,the most efficient cross-coupling catalysts feature the second- and the third-row transition metals,most notably palladium and rhodium,to achieve high turnover numbers.Despite the wide practicabilities of this methodologies,crosscoupling catalysis continues to attract significant interest,especially with respect to the development of more sustainable catalysts based on abundant first-row transition metals,particularly nickel catalysis.
3.3.Ru catalysis
In 2005,the ruthenium-catalyzed coupling reaction of aldimines bearing the 3-picolin-2-yl group with arylboronatesviachelation assistance strategy was reported by Jun and coworkers (Scheme 36) [114].The reaction carried out in a mixture solvent of 1,4-dioxane and acetone,using Ru3(CO)12as catalyst,methyl vinyl ketone as additive which can suppress the reduction of imines dramatically.Diverse functional arylboronates and aldehydes can be tolerated,furnishing the corresponding ketimines,which were readily converted to ketones by hydrolysis.
3.4.Ni catalysis
In 2015,Gu group described the nickelN-heterocyclic carbene catalyzed cross-coupling reaction of aryl aldehydes with boronic esters.This method provides a practical,neutral,and mild synthetic approach to aryl ketones,employing Ni(cod)2/IPr as the catalyst,hexafluoroacetone as a hydrogen acceptor (Scheme 37) [115].Different substituted aryl aldehydes and aryl boronic esters were well tolerated in this reaction,irrespective of the position and nature of substituted groups.However,aliphatic aldehydes could not deliver the corresponding ketones under the same reaction conditions.Notably,it was found that the transformations with boronic esters,bearing linear or branched substituents,proceeded smoothly to afford the aryl alkyl ketones in good yields.Moreover,a series of deuterium labeling experiments demonstrated that a nickel hydride species was involved in this reaction,C−H bond activation was the rate-determining step.

Scheme 36.Ru-catalyzed coupling of aldimines with arylboronates.
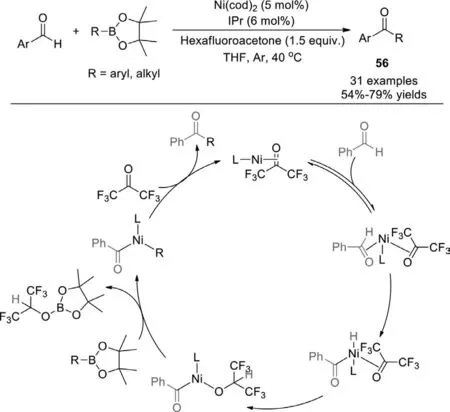
Scheme 37.Nickel N-heterocyclic carbene catalyzed cross-coupling of aryl aldehydes with boronic esters.
Through the similar reaction mechanism,several months later,Gu,Yuan and co-workers described the nickelN-heterocyclic carbene-catalyzed transformation of aromatic aldehydes into aryl ketones utilizing organozinc reagents [116].MgCl2was used as an additive,andp-methoxytrifluoroacetophenone was used as the hydrogen acceptor.A series of organozinc reagents and aryl aldehydes worked well,the corresponding aryl/aryl or alkyl/aryl ketones were achieved in good yields (Scheme 38).
4.Aldehyde as acyl radical
Transition metal-catalyzed C–H activation and radical reactions are two versatile strategies to construct diverse organic skeletons.Aldehydes are known to react with a variety of free radicals to form acyl radicals.And hydrogen atom abstraction from aldehydes by reactive oxygen radicals (e.g.,tBuO·) is known to be the cleanest way to generate acyl radicals.Due to the widespread applications of acyl radicals,coupled with the great value of ketones in organic synthetic chemistry,there have been a series of systematic studies providing methods for the carbonyl derivatization of aldehydes(Scheme 39).

Scheme 38.Nickel N-heterocyclic carbene-catalyzed cross-coupling reaction of aryl aldehydes with organozinc reagents.
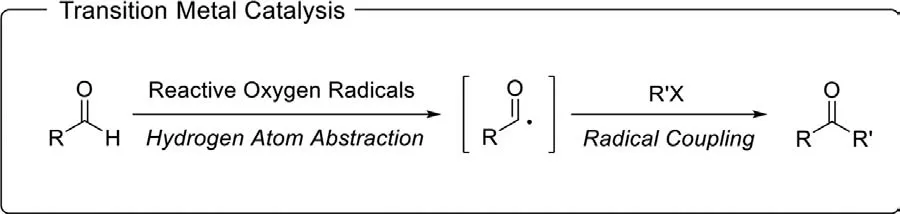
Scheme 39.Ketone synthesis via transition metal-catalyzed C–H activation and radical reactions.
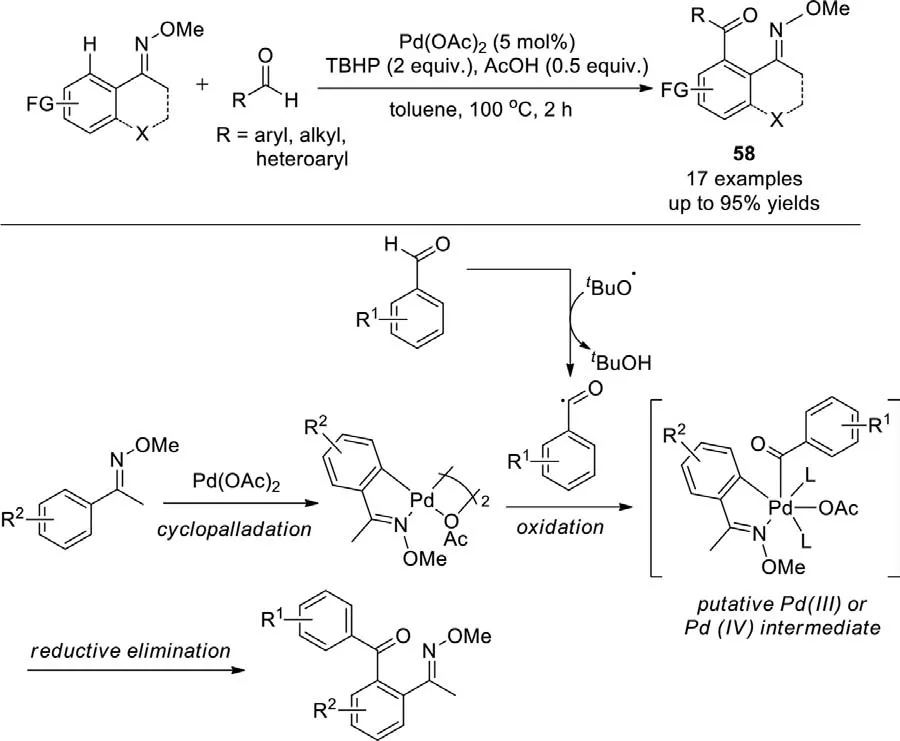
Scheme 40.Pd-catalyzed oxidative C-H bond coupling with aldehydes.
4.1.Pd catalysis
In 2010,Yu group developed the Pd-catalyzedortho-C-H acylation/cross coupling of aryl ketoneO-methyl oximes with aldehydes usingtert-butyl hydroperoxide (TBHP) as a source of reactive oxygen radicals (e.g.,tBuO·),which would act upon aldehyde to generate acyl radicalsin situ(Scheme 40) [117].With oximes as a directing group,the C–H activation is initiated byortho-selective cyclopalladation on the arene ring.The palladacycle would react with the acyl radicals to afford the product ketonesviaeither reactive Pd(IV) or dimeric Pd(III) intermediates.The coupling with aldehydes was achieved with remarkable regioselectivity,and both aliphatic and heteroaromatic aldehydes can be effectively coupled to the oximes.The acylation reactions constitute a versatile route to a diverse library of diaryl ketones,which are difficult to obtain by classical Friedel-Crafts acylation,and exhibit excellent functional group tolerance and regioselectivity.

Scheme 41.Pd-catalyzed direct acylation of aryl iodide with alkyl/aryl aldehydes.

Scheme 42.Pd-catalyzed remote aryldifluoroalkylation of alkenyl aldehydes.
In 2016,an environmentally benign strategy for the direct acylation of iodo arenes with alkyl/aryl aldehydes was established by Satyanarayana and co-workers,accessing to a wide variety of aryl−aryl and alkyl−aryl ketones in the presence of palladium catalyst (Scheme 41) [118].TBHP was used as oxidant to generate acyl radical and Ag2O facilitated the radical generation due to insoluble AgI formed in this biphasic system.This reaction,without activation of the carbonyl group and without directing group assistance,can be applied to the synthesis of antiplatelet drugnbutylphthalide (NBP) and the antispasmodic drug Pitofenone.
In 2017,Zhu group described the palladium-catalyzed threecomponent reaction between fluoroalkyl bromides,arylboronic acids,and alkenyl aldehydes,and provides facile access to 5-,6-,or 7-difluoroalkylated ketones under mild reaction conditions (Scheme 42) [119].The resultant products can be smoothly converted into CF2-containing tetrahydronaphthalenesviaa novel silver-catalyzed intramolecular decarboxylative cyclization of 5-aryl-2,2-difluoropentanoic acids.A radical mechanism is proposed for this palladium-catalyzed remote aryldifluoroalkylation.The reaction is initiated by a single-electron transfer (SET) between Pd(0) and the fluoroalkyl halides,thus producing radical species (62) with the concurrent formation of Pd(I).The addition of 62 across the C=C bonds of alkenyl aldehyde forms the fluoroalkyl radical 73,which can be converted into the acyl radical 64 through 1,n-HAT (n=5 or 6) from intramolecular aldehyde C−H bonds.The coupling of 64 with a Pd(I) species,followed by a transmetalation with ArB(OH)2,produces the intermediate 66.As a consequence of reductive elimination,the remote aryldifluoroalkylation products are formed,accompanied by the regeneration of Pd(0).
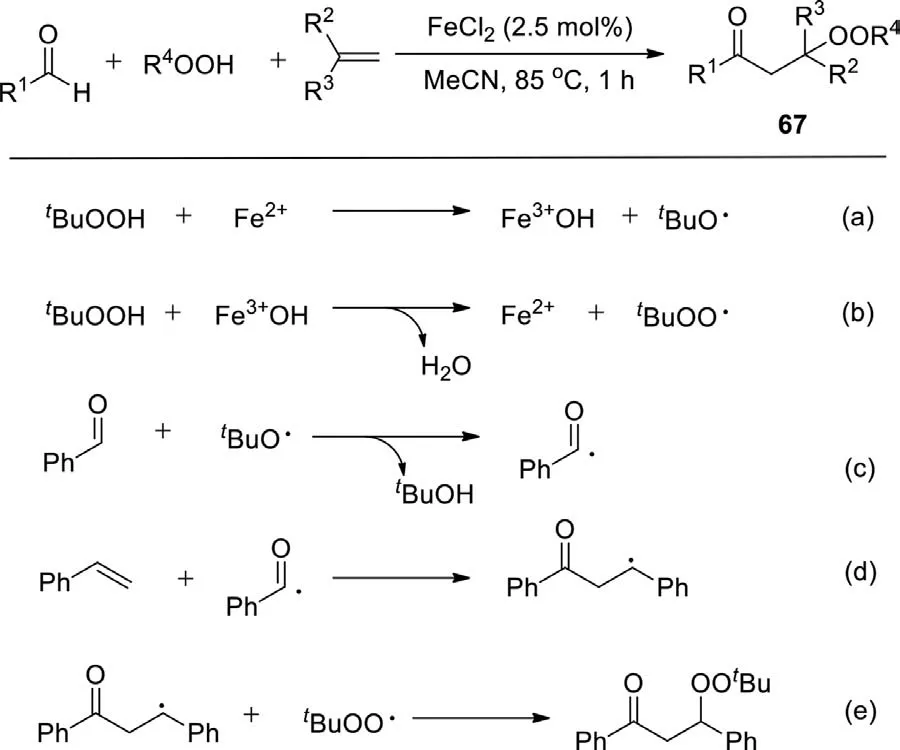
Scheme 43.Iron-catalyzed carbonylation-peroxidation of alkenes.
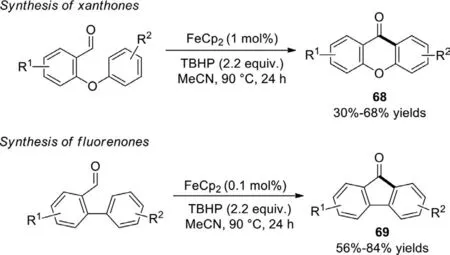
Scheme 44.Synthesis of fluorenones and xanthones via cross dehydrogenative coupling.
4.2.Fe catalysis
In 2011,Li group achieved the FeCl2-catalyzed three-component reaction of aldehydes,alkenes,and hydroperoxides to synthesizeβ-peroxy ketones (Scheme 43) [120].This three- component reaction can be also applied to the synthesis ofα-carbonyl epoxides,through either a stepwise base-induced epoxidation of the separatedβ-peroxy ketones or a one-pot process by simply adding base to the reaction mixture after the completion of the threecomponent reaction.Mechanistically,alkyloxy and alkylperoxy radicals are generated by iron catalyst.Subsequently,hydrogen atom abstraction bytert-butoxyl radical,gives the corresponding acyl radical.The radical addition followed by radical coupling leads to the final product.
In 2013,Studeret al.revealed a cross dehydrogenative coupling(CDC) of biphenyl-2-carboxyaldehyde to generate xanthones and fluorenones through base-promoted homolytic aromatic substitutions (BHASs),empolying TBHP as oxidant and FeCp2(0.1 mol% or 1 mol%) as radical initiator (Scheme 44) [121].The reaction starting with readily availableortho-formyl biphenylethers or biphenyls,was well tolerated to diverse substituents and provided the products in moderate to excellent yields.Mechanistically,the radical process was triggered by the generation of acyl radical from aldehyde in the presence oftBuOOH and Fe(II)X2.In addition,this work did not require any external strong base.
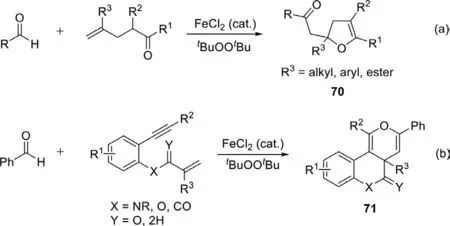
Scheme 45.Iron-catalyzed acylation-oxygenation of terminal alkenes and [2+2+2]annulation of benzene-linked 1,7-enynes with aldehydes.
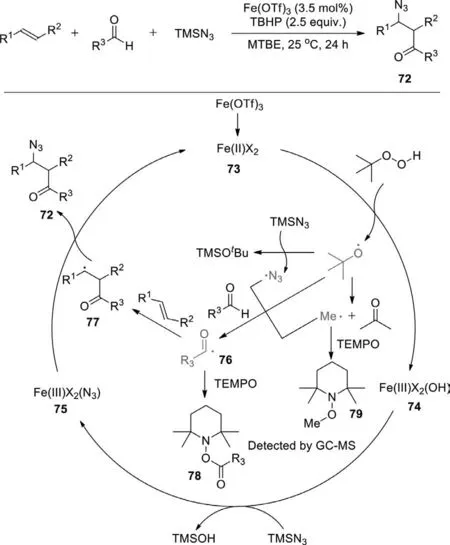
Scheme 46.Iron-catalyzed radical acyl-azidation of alkenes.
In 2015,Li group reported the iron-catalyzed acylationoxygenation of terminal alkenes using FeCl2as catalyst anddi-tertbutylperoxide as oxidant (Scheme 45a) [122].Acyl radicals generated by the oxidation of aldehydes add to terminal alkenes and followed by intramolecular oxygenation give functionalized 2,3-dihydrofuran derivatives bearing a quaternary carbon.Using the same reaction conditions,they developed an iron-catalyzed radical[2+2+2]annulation of benzene-linked 1,7-enynes with aldehydes in 2016 (Scheme 45b) [123].With this method,a variety of fused[6.6.6]pyran molecules are built in an efficient and selective manner.The aldehydic radical-mediated strategy exhibits a particularly attractive dual role,which triggers and terminates the domino cyclization.
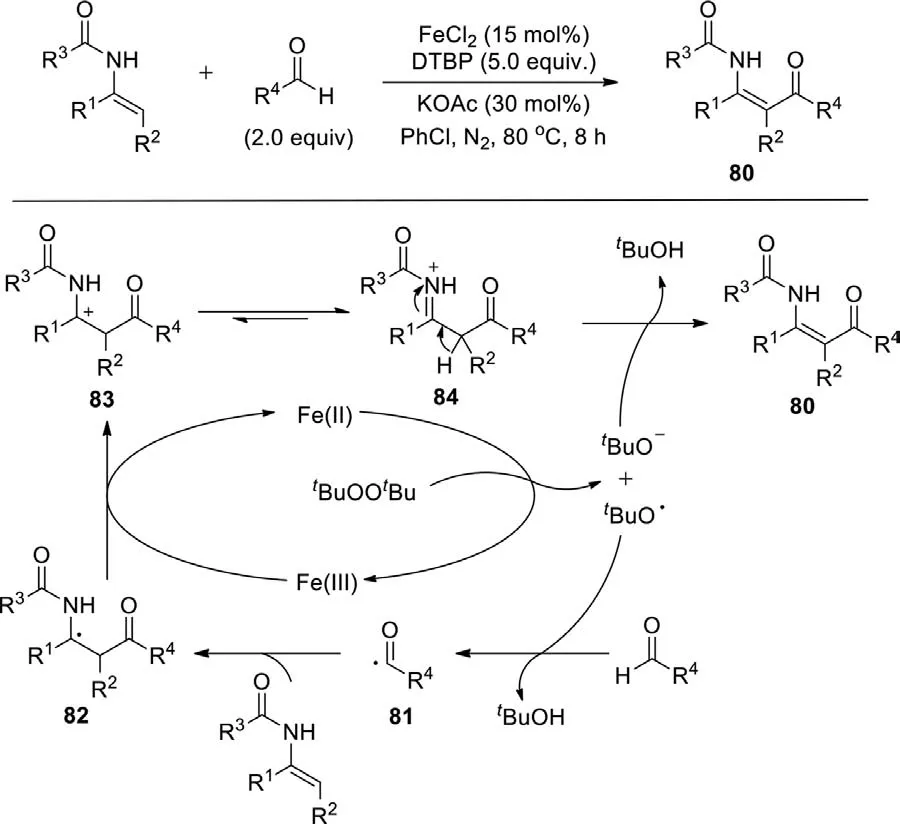
Scheme 47.selective dehydrogenative acylation of enamides with aldehydes leading to β-ketoenamides.
In 2019,Bao group developed the iron-catalyzed acyl-azidation of alkenes under mild reaction conditions (Scheme 46) [124].Aromatic or aliphatic aldehydes can be used as the acyl radical precursors;TMSN3is employed as the azido source;TBHP is the initiator.The synthesized unsymmetricalβ-azido ketones can be easily transformed into valuable functionalized compounds,such asγ-aminol,γ-azido alcohol,β-azido oxime,β-azido ester and triazoles.Initially,an iron(II) species (73) undergoes a SET process with TBHP to afford the iron(III) species (74) and atert-butyloxyl radical,which can decompose to a methyl radical or release an azido radical from TMSN3.Then,thetert-butyloxyl radical (or the methyl radical or the azido radical) abstracts an H-atom from the aldehyde to afford an acyl radical,which is trapped by an alkene to produce a more stable carbon-centered radical 77.Upon ligand exchange with TMSN3,the iron(III) species (74) is converted into an iron(III) species (75),which then undergoes azido group transfer with 77 to afford the acyl-azidation product,regenerating the iron catalyst.
In 2020,Hu group presented the iron-catalyzed dehydrogenative acylation of enamides with aldehydes to affordβketoenamides with broad substrate scope,good functional group compatibility,and exquisite stereoselectivity (Scheme 47) [125].The acyl radical 81 was producedviathe hydrogen-abstraction from the corresponding aldehyde by thetert-butoxyl radical,which was generatedviathe decomposition of DTBP by FeII.Subsequently,the acyl radical 81 attacked the enamide at theβ-position providing the intermediate 82,which underwent an oxidation by FeIIIto afford the tentative cationic species 83.83 was transformed the iminium intermediate 84,followed by deprotonation with the aid of thetert-butoxide anion to deliver the dehydrogenative acylation product.
4.3.Cu catalysis
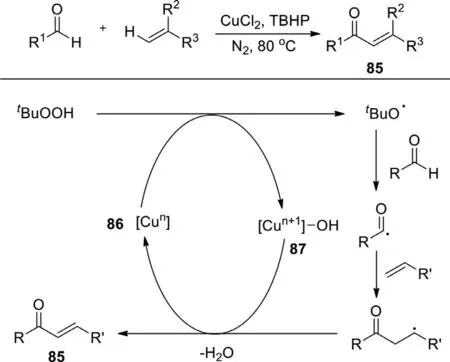
In 2013,Leiet al.reported the first copper-catalyzed oxidative coupling reaction of aldehydes with alkenes to formα,β-unsaturated ketone compounds using CuCl2as catalyst(Scheme 48) [126].Due to the unnecessary pre-functionalization of aldehydes and alkenes,this reaction is a very noticeable and superior protocol to synthesizeα,β-unsaturated ketones.The mechanism indicated that alkyloxy radical was formed from TBHP under the action of 86.Then aldehyde went through a hydrogen atom transfer (HAT) in the presence of the resulting alkyloxy radical to generate an acyl radical.Subsequently,the addition of acyl group to alkene provided benzylic radical,followed by oxidation and deprotonation to obtain the desired product 85 while regenerating 87.
Scheme 48.Copper-catalyzed oxidative coupling of aldehydes with alkenes to synthesize α,β-unsaturated ketones.
Scheme 49.Copper-catalyzed oxidative coupling of quinazoline 3-oxides with unactivated aldehydes.
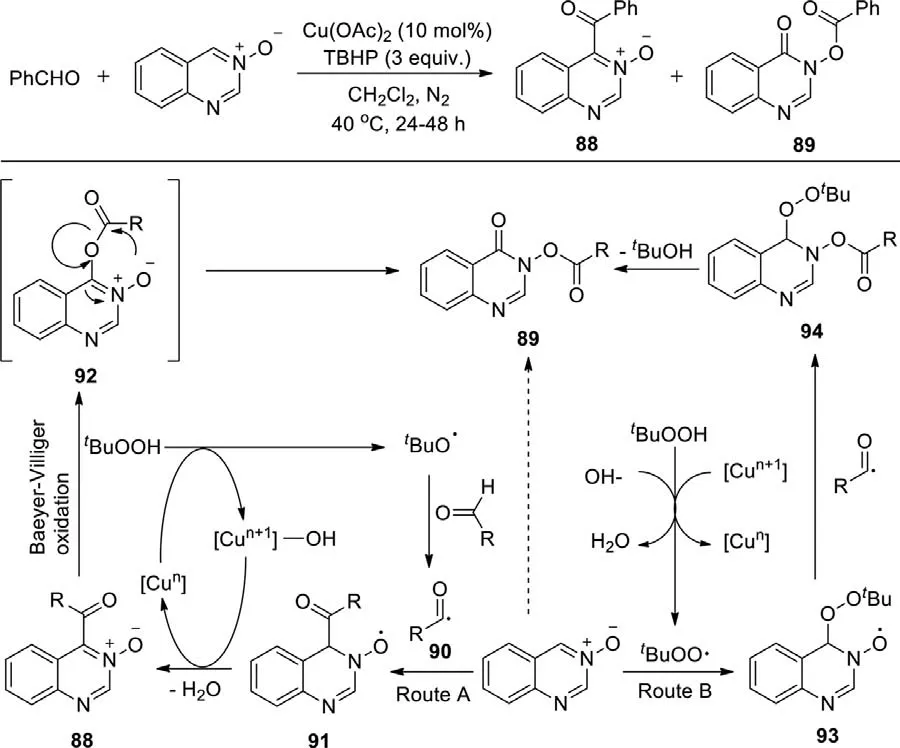
Owing to quinazoline derivatives are the core structure of many natural compounds and pharmaceuticals,the effective synthesis of these compounds is of great significance.In 2016,Wang and Zhaoet al.disclosed a copper-catalyzed oxidative coupling reaction of unactivated aldehydes with quinazoline 3-oxides,providing quinazoline ketones 88 followed by the direct formation of quinazoline esters 89 in one-pot reaction,using Cu(OAc)2as catalyst and TBHP as oxidant (Scheme 49) [127].The mechanism illustrated that the final product was formed by two routes: The alkyloxy radical from TBHP abstracted the hydrogen atom from aldehyde to provide the acyl radical 90,followed by the combination of quinazoline 3-oxide and acyl radical to afford the radical 91,which was further oxidized to form the ketone.After that,the ketone underwent the Baeyer–Villiger oxidation to deliver an intermediate 92,and cyclic hydroxamic ester 89 was furnished by the intramolecular acyl transfer (Route A).In another way,TBHP was dehydrogened to obtain atert-butyl peroxy radical,which reacted with quinazoline 3-oxide to give a radical 93,then 93 combined with the acyl radical to afford ester 94.At last,ester 94 was transformed into cyclic hydroxamic ester 89viafurther fragmentation (Route B).
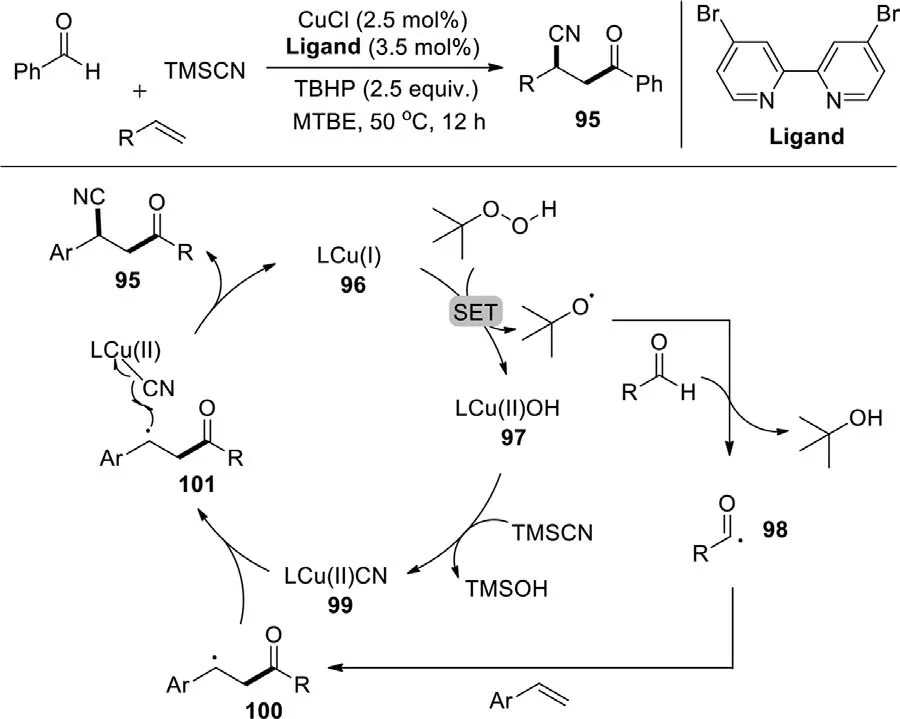
In 2019,Bao group reported the copper-catalyzed radical acylcyanation of alkenes with aldehydes to access various unsymmetricalβ-cyano ketones,employing TBHP as initiator (Scheme 50)[128].The ligated copper(I) catalyst (96) and TBHP undergoes a SET process to afford CuIIspecies (97) and OtBu radical.Then,the OtBu radical extracts a hydrogen atom from the aldehyde to propagate the acyl radical (98) and tert-butanol.Subsequently,the acyl radical captures a vinylarene to form a more stable benzyl radical (100).Upon ligand exchange with TMSCN,the CuIIspecies 97 converts to cyanocopper complex (99),which then undergo cyano group transfer with the benzyl radical 100 to access the acylcyanation product,meanwhile regenerating the copper(I) catalyst 96.
Scheme 50.Copper-catalyzed radical acyl-cyanation of alkenes.
Scheme 51.Synthesis of phosphonate chroman-4-ones through cascade radical cyclization-coupling of 2-(allyloxy)arylaldehydes.
4.4.Ag catalysis
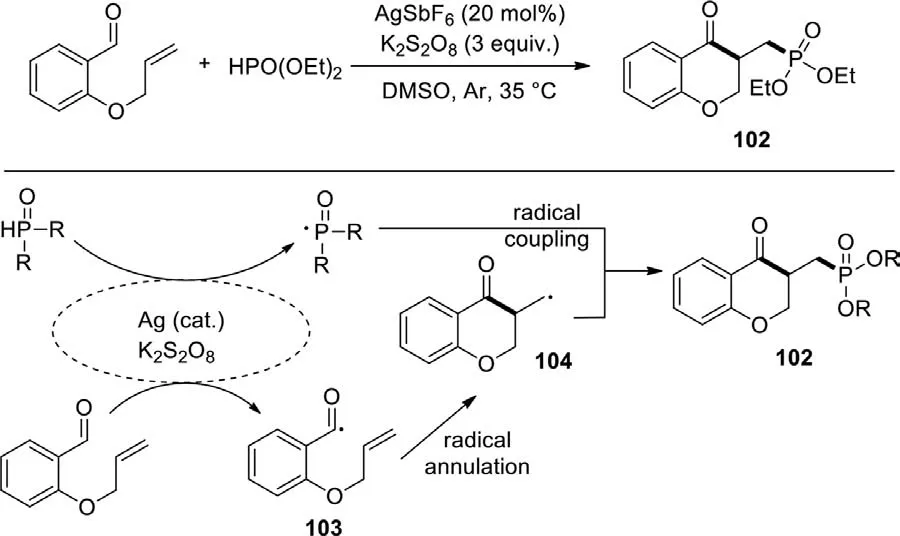
The functionalized chroman-4-one skeletons are widely used in biological and pharmaceutical compounds,therefore the development of an effective approach to synthesize such compounds has received much attention.In 2016,Li’s group reported a radical cyclization reaction of diethyl phosphite and 2-(allyloxy)-benzaldehyde to form phosphonate chroman-4-one derivatives under mild reaction conditions (Scheme 51) [129].This reaction proceeded successfully by using AgSbF6as catalyst and K2S2O8as oxidant.To explore the process of this reaction,two possible synthetic paths were proposed.Initially,an acyl radical and a phosphonyl radical were generated from aldehyde and diethyl phosphite respectively in the presence of silver catalyst and K2S2O8.The resulting acyl radical 103 underwent intramolecular cyclization to produce an alkyl radical 104.Subsequently,the coupling between the nucleophilic alkyl radical 104 and the electrophilic phosphonyl radical afforded the desired product 102.Pleasingly,the protocol also can be used for synthesizing azide and hydroxy functionalized chroman-4-ones.
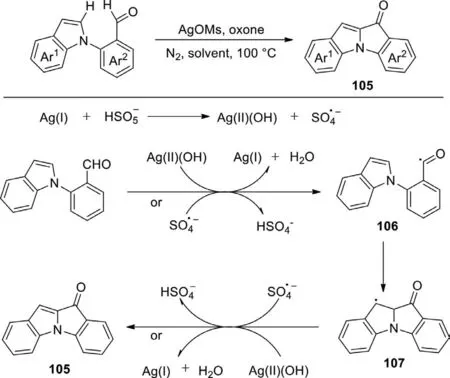
Scheme 52.Silver-catalyzed intramolecular C-2 selective acylation of indoles with aldehydes.
Indole-indolone compounds always occupy a significant position in the application of various biomedical products and pharmacological compounds.Although great achievements have been made in the development of transition metal-catalyzed direct activation of C−H bonds or the abstraction of hydrogen from the aldehyde,no progress has been made in the catalytic synthesis of indole-indolone.In 2016,Rao’s group reported a silvercatalyzed activation of C−H bond of aldehyde for the construction of indole-indolone compounds with good selectivity for the first time (Scheme 52) [130].The reaction occurred well in the presence of oxone as oxidant and AgOMs as the catalyst,which could obtain the product in good yields.The experiment on the mechanism was conducted.Initially,peroxymonosulfate anion disproportionates into a hydroxide anion and a sulfate radical anion in the presence of [Ag(I)]salt,meanwhile,the [Ag(I)]salt is oxidized to[Ag(II)]species.Then the acyl radical 106 was obtained via the oxidation of C-H bond of aldehyde by [Ag(II)]species or the abstraction of a hydrogen from aldehyde by sulfate radical anion.The acyl radical 106 underwent an intramolecular radical addition to afford the radical 107,which was subsequently oxidized by sulfate radical anion or [Ag(II)]species,and then underwent the deprotonation to generate the product 105.
4.5.Ni catalysis
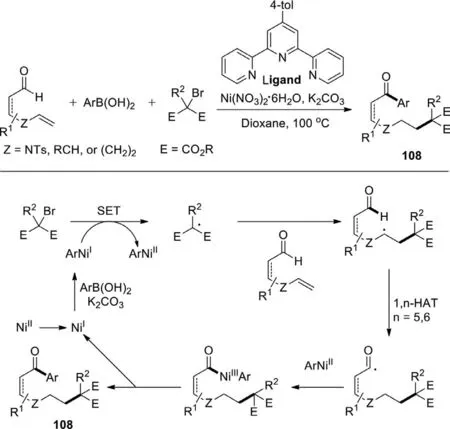
In 2018,Zhu group developed the nickel-catalyzed remote arylation of alkenyl aldehydes triggered by radical alkylation with tertiaryα-carbonyl alkyl bromides,thus producing a quaternary carbon center containing ketones in promising yields with broad functional group compatibility (Scheme 53) [131].Preliminary mechanistic studies suggest that the combination of a 1,n-HAT (n=5 or 6) from alkyl radicals to aldehyde C−H bonds with nickel catalysis may account for the realization of this reaction.
4.6.Mn catalysis
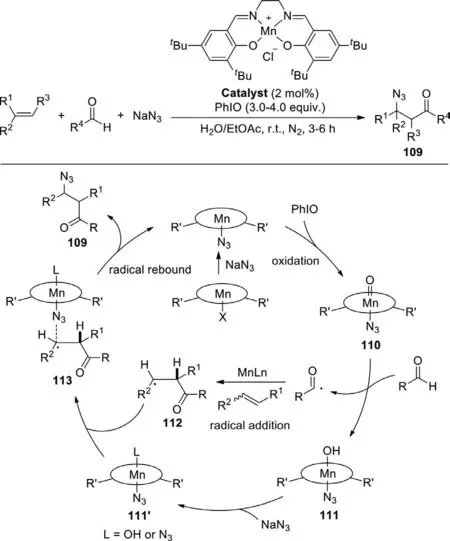
In 2018,Wanget al.describe the three-component reaction of aldehydes,olefins,and sodium azide for the installation of C=O and N3groups into the double bond based on(salen)Mn(III) catalysis system (Scheme 54) [132].In the conversion,the manganese-bound azide was oxidized by PhIO,generating the key reactive oxomanganese(V) intermediate 110,which is capable of abstracting hydrogen from the aldehyde to form intermediate 111 and an acyl radical.The subsequent acyl radical addition of a manganese-activated alkene would generate radical intermediate 112.Finally,azido-reboundviacomplex 113 released the final product of acylazidation.
Scheme 53.Nickel-catalyzed remote arylation of alkenyl aldehydes.
Scheme 54.(Salen)Mn(III)-catalyzed chemoselective acylazidation of olefins.
5.Aldehyde as carbon radical acceptor
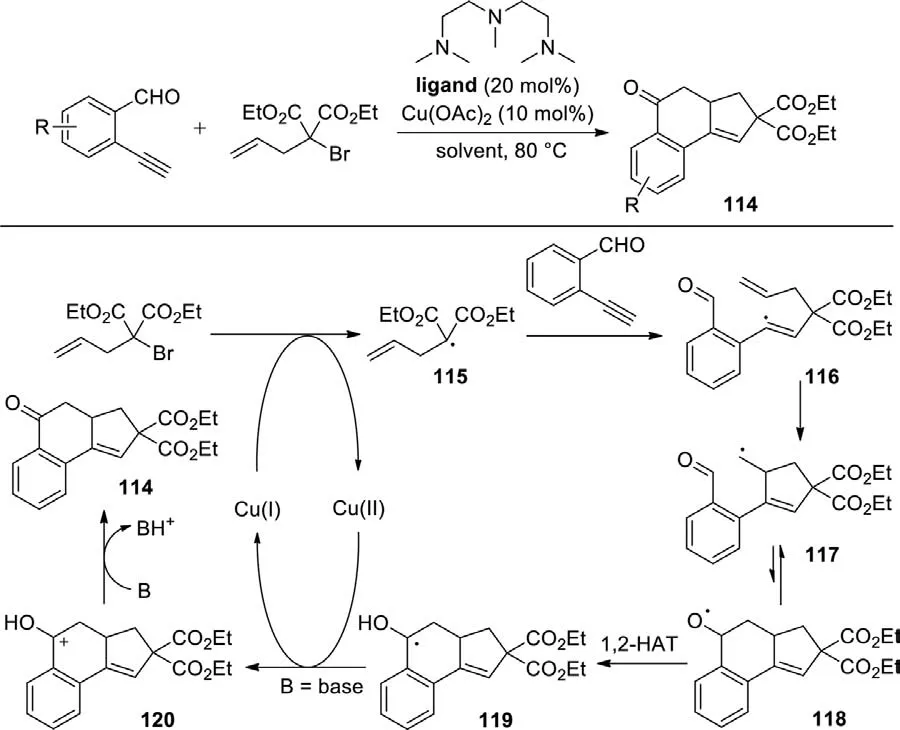
In the aforementioned work,aldehyde generally was used as a carbon radical donor to initiate ketone synthesis,while employing aldehyde as carbon radical receptor was difficult to achieve.In 2016,Zhu and co-workers developed the copper-catalyzed cascade annulation of enynals with alkenyl or alkynylα-bromocarbonyls,yielding various cyclohexenone-fused polycyclic compounds,using Cu(OAc)2as catalyst and pentamethyldiethylenetriamine (PMDETA)as ligand.Diethylazodicarboxylate (DEAD) was used as reductant forin situgeneration of the CuIcatalyst (Scheme 55) [133].
Scheme 55.Copper-catalyzed cascade annulation of enynals with unsaturated αbromocarbonyls.
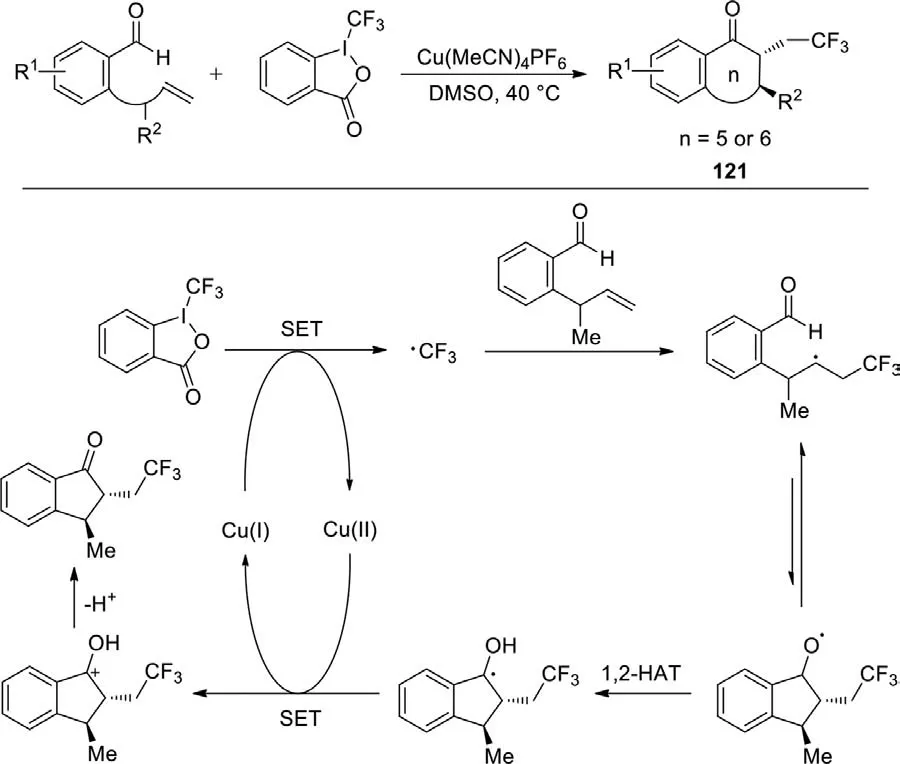
Scheme 56.Copper-catalyzed acyltrifluoromethylation of alkenes.
The mechanism showed that radical 115 and CuIIwere initially obtained by a single electron transfer (SET) between CuIandαbromocarbonyls,then the addition of the resulting radical to C≡C delivered alkenyl radical 116,which is converted to the alkyl radical species 117viaa 5-exo-trig cyclization.Then,an intramolecular addition of carbon radical to the aldehyde group led to the generation of alkoxy radical 118,which underwent 1,2-HAT to provide benzyl radical 119.Subsequently,the carbon-centered radical 119 was transformed to a cationic intermediate 120 by a second SET process with the aid of CuII,and CuIwas simultaneously regenerated.Finally,the intermediate 120 was deprotonated under the action of base to produce target product.Furthermore,the outstanding characteristics of this protocol were excellent step-economics.Up to six new C-C bonds and four new carbocycles can be established in a single reaction.
The trifluoromethylation of alkenes,which could straightforwardly construct the C-CF3bond,has received extensive attention because of its good step efficiency.In 2017,Zhuet al.firstly reported a trifluoromethylation of unactivated alkenes to form trifluoroethyl indanones and related cyclic ketones using Cu(MeCN)4PF6as catalyst (Scheme 56) [134].In this process,the addition of trifluoromethyl radical to alkenes generated the nucleophilic alkyl radical.Subsequently,an alkoxy radical was generated through intramolecular cyclization.And then 1,2-HAT,SET,and deprotonation proceeded successively to provide the final product.This method made a significant contribution to drug application and development through the combination of CF3group and an indanone molecule.
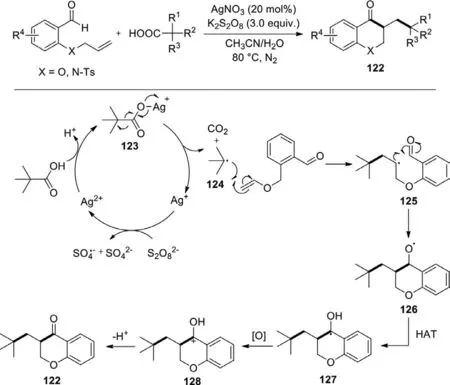
Scheme 57.Silver-catalyzed decarboxylative cascade radical cyclization of tertcarboxylic acids and o-(allyloxy)arylaldehydes.
Efficient synthetic protocols of chroman-4-one derivatives have gotten widespread development during the past decades owing to their special physiological and biological properties.The cascade radical cyclization ofo-(allyloxy)arylaldehydes with different radicals towards the formation of diverse chroman-4-one derivatives has been rapidly developing.
Zhu group disclosed the silver nitrate-catalyzed oxidative radical decarboxylative cyclization reaction ofo-(allyloxy)arylaldehydes and tertiary carboxylic acids to obtain 3-alkyl-substituted chroman-4-one derivatives under mild conditions (Scheme 57)[135].The substrate scope of this reaction was inspected,indicating that variouso-(allyloxy)arylaldehydes and tertiary carboxylic acids with different substituents were well tolerated and generated corresponding products with moderate to good yields.A proposed mechanism illustrated that Ag(I) was firstly transformed into Ag(II) in the presence of persulfate,then Ag(II) combined with tertiary carboxylic acid to produce silver complex 123 which was converted into a radical 124 and regenerated Ag(I) through a single electron transfer,meanwhile CO2was released.Subsequently,the direct addition of radical 124 to the C=C bond ofo-(allyloxy)arylaldehyde afforded the radical intermediate 125,which successively underwent an intramolecular cyclization,a HAT and oxidation to deliver an intermediate 128.Finally,expected product 122 was obtained by the deprotonation of intermediate 128.
The utilization of cheap and easily obtainable carboxylic acids as raw materials has great advantages,so that the decarboxylation cross-coupling reaction for the construction of C-C and C-X bonds has received considerable attention in the field of synthesis.Enlightened by previous works,in 2017,Wuet al.firstly reported a silver nitrate-catalyzed cascade decarboxylation radical addition/cyclization reaction which performed smoothly betweenα-oxocarboxylic acids and aromatic aldehydes (Scheme 58) [136].Eventually,it constructed dihydroflavonoid derivatives with good efficiency and broad substrate scope upon the oxidation of ammonium persulfate.In addition,diverseα-oxocarboxylic acids and 2-(allyloxy)benzaldehyde derivatives bearing different substituents were investigated in this work,and the desirable products exhibited good yields.A feasible mechanism showed that the aldehyde acted as a carbon radical acceptor in this reaction.Initially,the carboxylic acid underwent decarboxylation under the action of silver and persulfate anion to give an acyl radical 130,then the addition of 130 to the C=C bond of the aldehyde formed radical 131,which afforded an alkoxyl radical 132 by intramolecular cyclization,followed by 1,2-HAT to produce a benzyl radical 133.Subsequently,the resulting benzyl radical went through dehydrogenation to generate the final product 129.
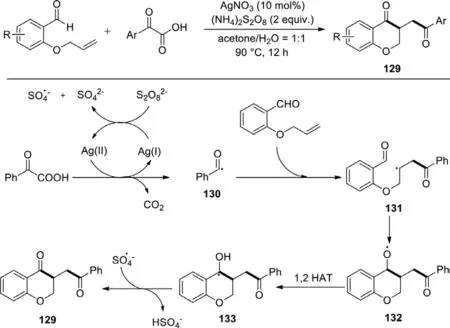
Scheme 58.Silver nitrate-catalyzed cascade decarboxylation and oxidative cyclization.
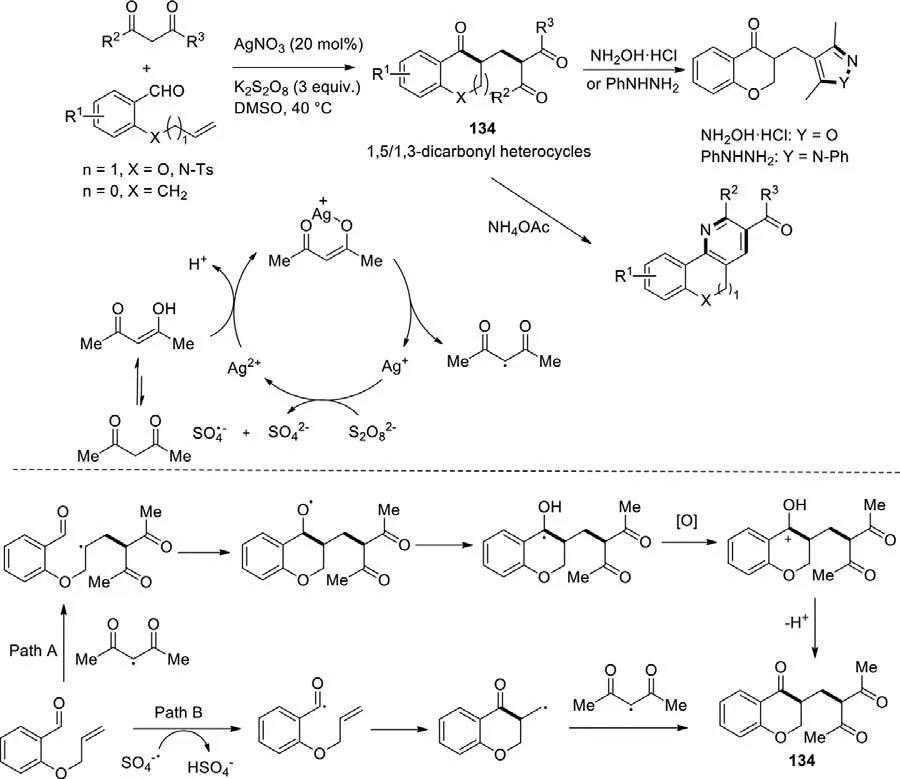
Scheme 59.Silver-catalyzed radical cascade cyclization toward 1,5-/1,3-dicarbonyl heterocycles.
It is well known that dicarbonyl compounds are a sort of important feedstock for the synthesis of various heterocycles.In the past several years,many efforts have been devoted to developing radical cascade reactions to efficiently afford the dicarbonyl heterocycles.A relatively convenient silver-catalyzed radical cascade cyclization was reported by Chen and Yuet al.in 2018 (Scheme 59) [137].In the presence of AgNO3and K2S2O8,multifarious 2-functionalized benzaldehydes reacted with 1,3-dicarbonyl compounds to form 1,5- or 1,3-dicarbonyl heterocyclic compounds including chroman-4-one,indanone or 2,3-dihydroquinoline-4(1H)-one.After that,the generated 1,5-/1,3-dicarbonyl heterocycles could react with NH4OAc,NH2OH·HCl,or PhNHNH2respectively,and finally produced polycyclic compounds with diverse structures.This is the first example of constructing a large variety of 1,5-/1,3-dicarbonyl heterocyclesviaa silver-catalyzed radical cascade cyclization reaction by directly using 1,3-dicarbonyl compounds as alkyl radical precursors.
Based on the foundation about cascade radical annulation ofo-(allyloxy)arylaldehydes with different radicals for the preparation of diverse chroman-4-one derivatives,Liu’s group reported a novel access to chroman-4-one derivatives by silver-catalyzed cascade radical cyclization ofo-(allyloxy)arylaldehydes with cyclopropanols in 2019 (Scheme 60) [138].Under the mild oxidative conditions,various cyclopropanols ando-(allyloxy)arylaldehyde derivatives were explored and rewarded good yields.
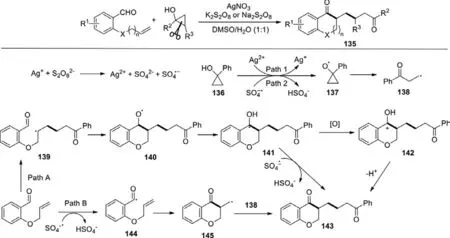
Scheme 60.Silver-catalyzed radical cyclization of 2-(allyloxy)arylaldehydes with cyclopropanols.
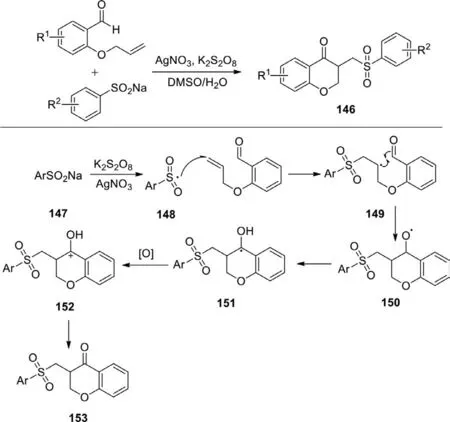
Scheme 61.Silver-catalyzed cascade radical cyclization of sodium sulfinates and o-(allyloxy)arylaldehydes.
Two plausible mechanisms were elucidated by several control experiments.In path A,o-(allyloxy)benzaldehyde reacted withβketo radical 138 whichin situgenerated from the oxidation and rearrangement of cyclopropanol 136 to form the radical intermediate 139,followed by cyclization to afford an oxygen radical 140.Then the benzyl radical 141 was formed through a 1,2-HAT process.Finally,the further oxidation and deprotonation yielded the desired product 143.Alternatively,in path B,sulfate radical anion abstracted the C-H bond of aldehyde,and generated acyl radical 144.Subsequently the acyl radical 144 underwent 6-exo-trig cyclization to provide the radical intermediate 145,which reacted with radical 138 to generate 143.
In 2019,Wang and co-workers conducted a similar exploration about the synthesis of chroman-4-one and indanone derivatives through radical cascade ring opening/coupling/cyclization [139].In addition to the synthesis of chroman-4-one,the indanone derivatives were also successfully prepared in good yields.In terms of the reaction mechanism,different opinions arise from the step that the hydrogen of the benzyl radical 141 was abstracted by the sulfate radical to afford the final products.
In 2020,Wang’s group reported silver-catalyzed cascade radical cyclization ofo-(allyloxy)arylaldehydes with sodium sulfinates to synthesize functionalized chroman-4-ones (Scheme 61) [140].Under the optimized reaction conditions that using K2S2O8as oxidant and AgNO3as the catalyst,this reaction displayed a wide substrate scope to yield the desired products in moderate to good yields.In this procedure,o-(allyloxy)benzaldehyde reacted with sulfonyl radical 148 which was generated from the sodium benzenesulfinate in the presence of K2S2O8and AgNO3to produce 149.Followed by intramolecular cyclization and HAT,it afforded the intermediate 151.After the oxidation and deprotonation of 151,the desired product 153 was obtained.This efficient method for the synthesis of functionalized chroman-4-ones derivatives greatly expands the focus on developing economical,practical and environmentally benign protocols for preparation of chroman-4-ones.
6.Conclusions
Ketones have been standing at the center of stage in synthetic organic chemistry,due to their versatile applications in the synthesis of fine chemicals,pharmaceuticals,and agrochemicals,and are already key players in chemical industries and pharmaceutical industries.Over the past few decades,transition metal-catalyzed direct conversions of aldehydes to ketones have drawn great attention.These transformations,in easy and straightforward manners,have excellent practical value,and broad application prospects.Herein,we have provided a comprehensive overview of the historic advancements starting from 2000,and mechanistic details of various aldehydes to generate corresponding ketones under different reaction conditions.We summarize these transformations based on the four major mechanisms: (1) Carbonyl-Heck reaction,that is 1,2-insertion of organometal species to aldehydic C=O double bond,(2) Direct insertion of transition metal catalysts to aldehydic C−H bond,(3) Aldehyde as acyl radical,(4) Aldehyde as carbon radical acceptor.We hope this systematic overview of ketone synthesis employing aldehydes as starting materials will serve as a useful reference for organic chemists and stimulate further innovations in this exciting research field.We are convinced that in the near future the application of these powerful methodologies will accelerate further,not only in academic research but also in industry.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful for the generous financial support from the National Natural Science Foundation of China (No.21801206),the Chunhui Program of Ministry of Education of China (No.5180210003),Program for Young Talents of Shaanxi Province (No.5113190023),the Joint Research Funds of Department of Science&Technology of Shannxi Province and Northwestern Polytechnical University (No.2020GXLH-Z-015),the Science,Technology and Innovation Commission of Shenzhen (No.JCYJ20190809160211372),the Ao’xiang Overseas Scholars Program of NPU and the Fundamental Research Funds for the Central Universities.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis