
C–F bond functionalizations of trifluoromethyl groups via radical intermediates
2022-06-18 10:52TesfayeTebekaSimurTianYeYouJieYuFengLianZhangYiFengWang
Chinese Chemical Letters 2022年3期
Tesfaye Tebeka Simur,Tian Ye,You-Jie Yu,Feng-Lian Zhang,Yi-Feng Wang
Department of Chemistry,University of Science and Technology of China,Hefei 230026,China
Keywords:C–F bond functionalizations Radical intermediates Trifluoromethyl groups Difluorinated compounds Monofluorinated compounds
ABSTRACT Selective functionalization of C–F bonds in trifluoromethyl groups has recently received a growing interest,as it offers atom- and step-economic pathways to access highly valuable mono- and difluoroalkylsubstituted organic molecules using simple and inexpensive trifluoromethyl sources as the starting materials.In this regard,impressive progress has been made on the defluorinative functionalization reactions that proceed via radical intermediates.Nevertheless,it is still a great challenge to precisely control the defluorination process,due to the continuous decrease of the C–F bond strength after the replacement of one or two fluorine atoms with various functionalities.This review article is aimed to provide a brief overview of recently reported methods used to functionalize C–F bonds of CF3 groups via radical intermediates.An emphasis is placed on the discussion of mechanistic aspects and synthetic applications
1.Introduction
Monofluoro- and difluoro-containing organic molecules have become tremendously important in pharmaceuticals [1,2],agrochemicals [3],and materials [4],owing to the unique properties of fluorine atom and its incorporation enhances the chemical and biological properties of the target compounds [5,6].In the last decades,numerous mono- and difluoroalkylating precursors and C–F bond formation strategies have been established for the introduction of fluorine atoms into organic compounds [7-11].Aside from this,the defluorinative functionalization of inert C-F bonds of CF3compounds is also an important pathway to the synthesis of useful partially fluorinated organic molecules [12-14].Such strategy has gained an ever-increasing interest,given the low cost of many CF3sources and numerous routes available to install CF3motifs [15,16].In general,the cleavage of C–F bond of CF3groups proceeds through heterolytic pathway,affording difluoro-substituted carbon cations or anions as intermediates,and a number of comprehensive reviews have summarized the progress of this research topic [6].
Defluorination reactionsviaradical intermediates represent a class of powerful transformations,in which the radical species can undergo different reaction pathways as compared to ionic intermediates,and thus providing versatile routes for chemical bond formation.However,such reactions are still insufficiently studied because of the high bond dissociated energy (BDE) of C–F bond that makes homolytic cleavage extremely difficult [17].Moreover,since C–F bond strength continuously decreases as defluorination proceeds,selectively formation of di- and monofluoroalkyl radical intermediates in a controlled manner becomes exceedingly diffi-cult and exhaustive defluorination is often resulted [18,19].So far,there have been some reports on deflurinative generation of radical interemediates and the CF3group is required to attached to aπ-system,such as arenes,alkenes,and carbonyls,so that the substrates can accept a single electron or a radical species and then induces fluoride anion elimination.This review will summarize recent progress of this topic and focus more on the mechanistic discussion.Meanwhile,the synthetic applications of the resulting radical intermediates will also be introduced.
2.C–F bond functionalization of trifluoromethylarenes
Selective functionalization of C(sp3)−F bond in trifluoromethylarenes has found an important place in modern organic synthesis,which provides direct access to the synthesis of a diverse range of aryldifluoromethyl and arylmonofluoromethyl molecules[14,20,21].Generally,defluorination occurs with the aid of UV irradiation [22,23]or under reductive reaction conditions.In the later cases,electrochemical reduction [24-26]and the use of Mg metal as the reducing agents are required [27-29].The reduction mechanismviaradical intermediates is shown in Scheme 1.The reaction begins with single-electron reduction of trifluoromethylarenes to generate radical anion intermediates,and subsequent elimination of a fluoride anion gives radical intermediates.These radical species can be easily reduced under strong reductive reaction conditions,affording carbanion intermediates,which can be trapped by electrophiles to deliver difluoro products.
For example,in 2017,Prakash and co-workers demonstrated a magnesium metal-promoted defluorination of bis (trifluoromethyl)arenes in the presence of Brønsted acid for the synthesis of difluoromethyl-containing arenes (Scheme 2) [30].In this protocol,functional groups like free amine,alcohol are well tolerated.However,reduction of all three C–F bonds was observed in the case of nitrile substituents.
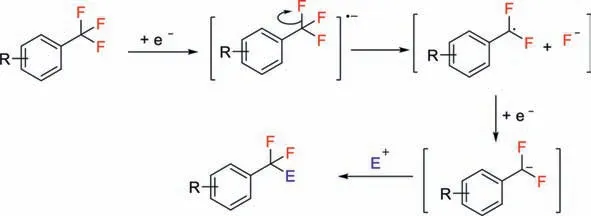
Scheme 1.A general mechanism for C–F bond cleavage of trifluoromethylarenes.

Scheme 2.Mg-promoted reductive defluorination of trifluoromethylarenes.
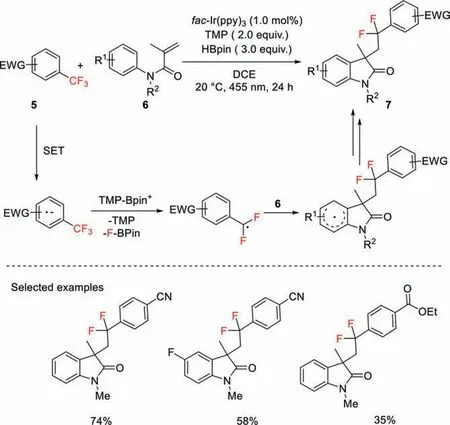
Scheme 3.C−F bond functionalization of trifluoromethylarenes through cascade radical addition with methylacrylamides.

Scheme 4.C(sp3)–F and C–C bond functionalization through defluoroalkylation–distal heteroaryl migration.
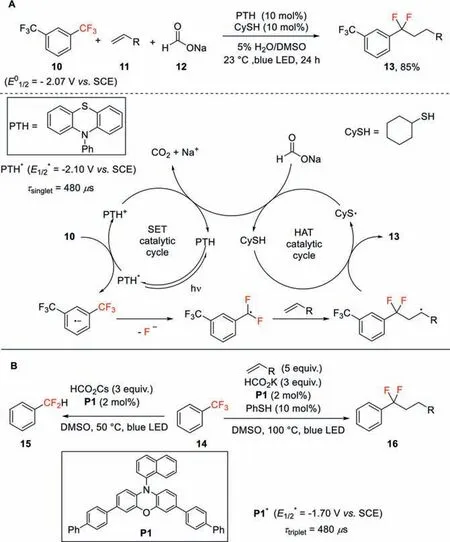
Scheme 5.A single C–F bond functionalization of ArCF3 via photoredox HAT dual catalytic strategy.
Scheme 6.Hydrodefluorination of trifluoromethylarenes.
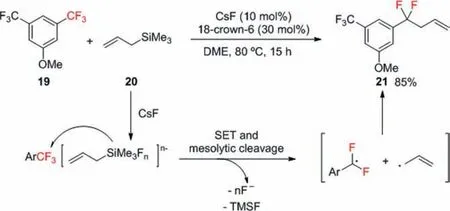
Scheme 7.C–F bond functionalization via fluoride-initiated sequential allylation
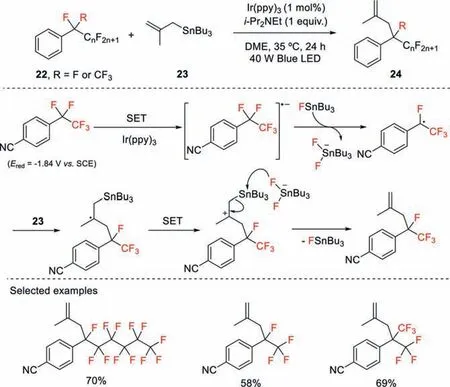
Scheme 8.C–F bond functionalization of perfluoroalkylarenes via defluorinative allylation reaction.

Scheme 9.The general mechanism of photoredox-catalyzed C–F bond cleavage of trifluoromethyl alkenes.
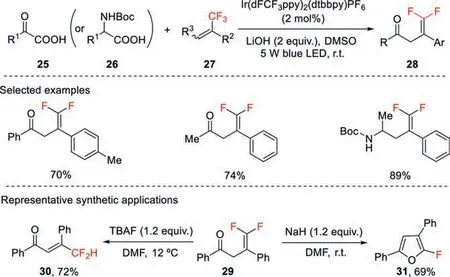
Scheme 10.Photocatalytic decarboxylative/defluorinative functionalization of trifluoromethyl alkenes.
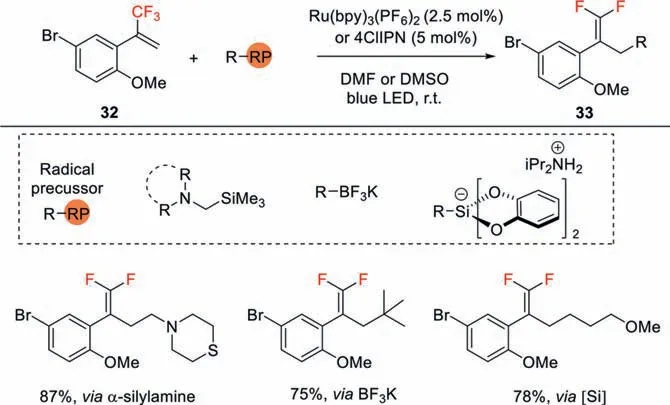
Scheme 11.Photocatalytic construction of 1,1-difluoroalkenes from different radical precursors.
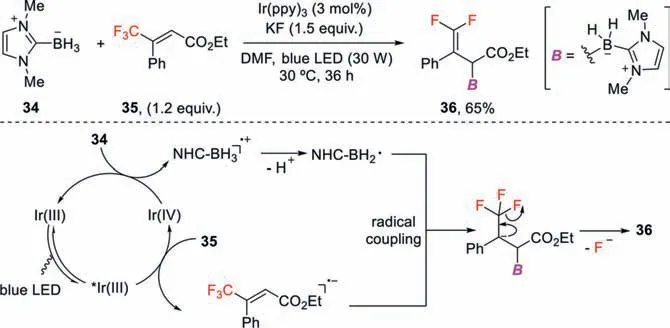
Scheme 12.Photoredox-catalyzed radical defluorinative borylation of trifluoromethyl alkenes.
The strategy shown above is difficult to capture the radical intermediate by various radical traps,as it prefers to undergo further reduction under those strong reductive reaction conditions.To address this challenge,developing new reductive protocols that can prevent the second reduction is desirable.Photoredox catalysis has recently emerged as a powerful method in organic synthesis.Notably,a large number of photoredox catalysts with a broad range of redox potentials are readily available,thus offering ample opportunities to precisely control the redox process.By taking this advantage,some defluorination reactions that can selectively generate difluoromethyl radical without further reduction have been reported.These radicals further participate in various transformations to access valuable ArCF2R derivatives.
Gschwind and König reported a protocol that emerges photoredox catalysis and Lewis acid activation,by which a single C−F bond of trifluoromethylarenes was selectively cleaved,giving aryldifluoromethyl radical intermediates.Those radicals were capable of performing radical addition to methacrylamides followed by cyclization to afford aryldifluoromethyl-tethered indolinone derivatives.Mechanistic studies suggested that thein situgenerated acidic borenium cation serves as an efficient fluoride scavenger that can accelerate the radical generation (Scheme 3) [31].
After that,Qiu and Guo have extended this strategy to enable a tandem C(sp3)–F and C–C bond functionalization through defluoroalkylation–distal migration of the heteroaryl group(Scheme 4) [32].The reaction begins with the reductive generation ofα,α-difluorobenzylic radical intermediates,which are then trapped by simple olefins.The resulting alkyl radical attacks an intramolecular heteroaryl ring to trigger a distal aryl migration,and the ensuing oxidation and deprotonation affords ketone products.
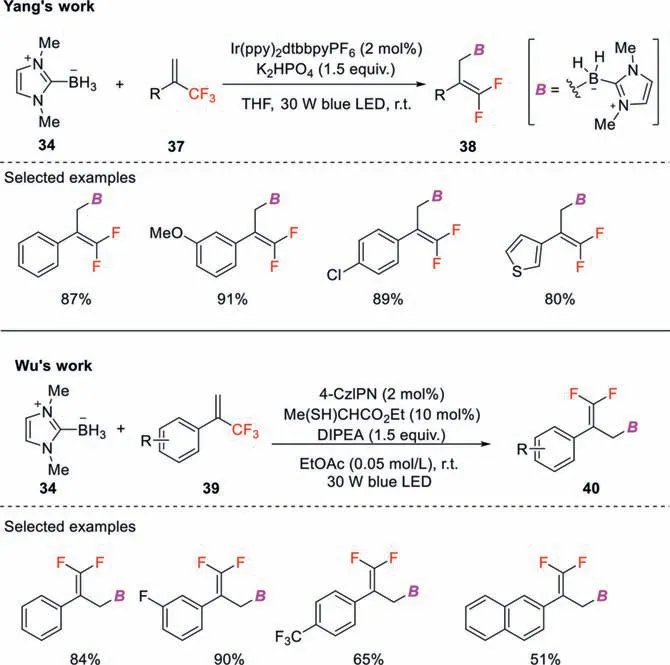
Scheme 13.Visible-light induced selective defluoroborylation of trifluoromethylalkenes.

Scheme 14.Mg metal-promoted C–F bond activation of trifluoromethyl carbonyls compounds.
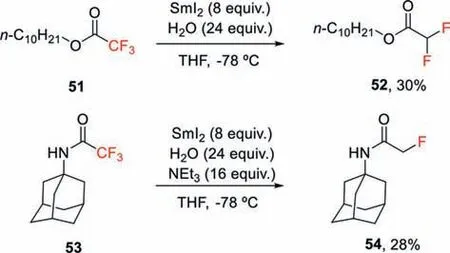
Scheme 15.SmI2-promoted defluorination of trifluoroacetyl esters and amides.
Jui and co-workers developed a new catalytic system for the single C–F bond cleavage of trifluoromethylarenes under visible light irradiation [33].In this protocol,N-phenylphenothiazine (PTH)was employed as photocatalyst and cyclohexanethiol (CySH) was used to promote hydrogen atom transfer (HAT).Under irradiation by blue LED,the highly reducing excited state PTH∗(E1/2∗=−2.10 Vvs.SCE) can deliver an electron to 1,3-bistrifluoromethylbenzene(E01/2=−2.07 Vvs.SCE) to generate difluorobenzylic radicals with the elimination of a fluoride anion.These radical intermediates then undergo efficient intermolecular coupling with simple alkenes to forge the desired difluoro products (Scheme 5A).This protocol is highly sensitive to the electronic properties of the trifluoromethylaromatic ring.Only the aryl ring bearing an additional strong electron-withdrawing group,such as CF3,phosphonate,and sulfonamide,is amenable in the reaction.
To address this limitation,the same group then developed a method employing Miyake’s phenoxazine (E1/2∗=−1.70 Vvs.SCE)as the photocatalyst,which has a long-lived triplet excited state.This method allows monodefluoroalkylation and monohydrodefluorination of unactivated trifluorotoluene derivatives (Scheme 5B)[34].As reported by the recent work of this group,a powerful single electron reductant (CO2•−) (E1/2∗=−2.20 Vvs.SCE) can be generatedviahydrogen atom transfer from a formate salt to a thiyl radical under the same reaction condition [35]and this reductant is likely also involved in this transformation.
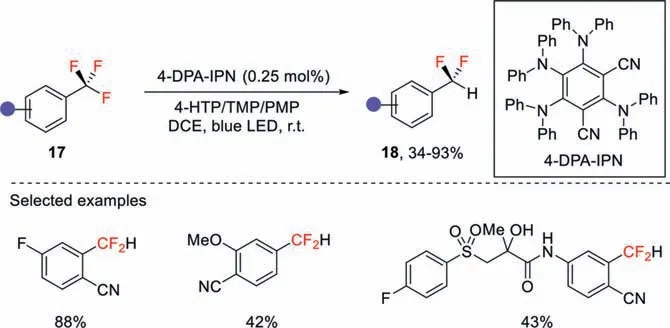
Gouverneur and co-workers quite recently disclosed a phtoredox protocol for the reductive defluorination of electron-poor trifluoromethylarenes under basic conditions (Scheme 6) [36].In this method,2,4,5,6-tetrakis(diphenylamino)isophthalonitrile (4-DPAIPN) was used as the organophotocatalyst,4-hydroxythiophenol was used as the hydrogen atom donor,under blue light irradiation,a series of complex trifluoromethylated drugs could be transformed into the corresponding difluoromethyl derivatives.
Bander and co-workers developed a fluoride-initiated coupling reaction between trifluoromethylarenes and allylsilanes to access allylatedα,α-difluorobenzylic compounds (Scheme 7) [37].First,fluoride ion act as Lewis base and coordinate to allyltrimethylsilane to give pentacoordinate and hexacoordinate silicate intermediates,which then undergo single electron transfer (SET) to the trifluoromethylarene.The following cleavage of both a C–F and a C–Si bond induces concurrent generation of anα,α-difluorobenzylic radical and an allyl radical,and the quick recombination affords a defluoroallylation product.The expelled fluoride anion then activates another molecule of allyltrimethylsilane.
Quite recently,Yasuda and co-workers reported a defluoroallylation reaction of perfluoroalkylarenes using Ir(III) photocatalyst and organotin reagent in cooperative mode of catalysis under visible light irradiation [38].In this transformation,the C–F bond functionalization takes place selectively at the benzylic position through perfluoroalkyl radicals generated from perfluoroalkylarenes by excited Ir(ppy)3in a single electron transfer pathway(Scheme 8).It should be noted that the destabilization and steric hindrance effects of the resulting perfluoroalkyl radicals are unfavorable for the sequential bond-forming reaction,thereby resulting in a retroprocess including back electron transfer and F−addition.Further DFT calculations suggest that thein situgenerated Bu3SnF is capable of trapping F−,which can suppress this retroreaction step.The generated perfluoroalkyl radicals then undergo addition to allylic stannanes followed by single electron oxidation and elimination of the stannyl cation,affording the corresponding defluoroallylation products.
3.C–F bond functionalizations of trifluoromethyl alkenes
Trifluoromethyl alkenes are privileged structural motifs for synthesizing a diverse range of partially fluorinated or nonfluorinated compounds.Over the past decades,various research groups have used visible-light-mediated reactions of carbon and heteroatom nucleophiles withα-trifluoromethyl alkenes to synthesizegem-difluoroalkenes [14,21].The general mechanism is shown in Scheme 9.Initially,the excited photocatalyst (PC∗) is reductively quenched by a radical precursor,affording the radical R•and PC•−.Radical addition to trifluoromethyl alkene formsα-CF3alkyl radical,which would be further reduced by PC•−to give sp3-hybridized carbanion and regenerate PC.Finally,β-fluoride elimination shifts the double bond to givegem-difluoroalkene products

Scheme 16.Sequential C–F bond functionalizations of trifluoroacetamides and trifluoroacetates via spin-center shifts.
This strategy was applied to the decarboxylative/defluorinative cross coupling ofα-keto acids and trifluoromethyl alkenes for the synthesis ofγ,γ-difluoroallylic ketones using an Ir-based photocatalyst excited under blue light (Scheme 10) [39].This reaction features mild reaction conditions,simple operation,and good functional group tolerance.The process could also be extended toN-Boc protectedα-amino acids for the synthesis of 1,1-difluorohomoallylic amines.The resulting functionalizedgemdifluoroalkenes can be transformed to various difluoromethylated compounds and monofluorinated heterocycles.
In 2017,Molander also demonstrated a visible light-mediated process for the synthesis of 1,1-difluoroalkenes by using an array of CF3-substituted alkenes with different carbon-radical precursorsviaradical defluorinative alkylation process (Scheme 11)[40].Whenα-silylamines were employed,various amine-tetheredgem-difluoroalkenes were produced.Potassium organotrifluoroborates and alkylbis(catecholato)silicates are also used as competent precursors of carbon centered radicals,providing a route to install a variety of alkyl-substitutedgem-difluoroalkenes.
Organoboron compounds have broad applications in chemical synthesis,material sciences,and medicinal chemistry.Recently,the groups of Wang [41],Yang [42]and Wu [43]independently reported photoredox catalysis-enabled radical defluorinative borylations of trifluoromethyl alkenes to afford a wide range ofgemdifluoroallylboranes.In Wang’s work (Scheme 12),the key step is the generation ofN-heterocyclic carbene (NHC)-BH2•viaa singleelectron oxidation of NHC-BH3(Ep/2=+0.76 Vvs.SCE) by IrIV(ppy)3(E1/2red[IrIV/IrIII]=+0.77 Vvs.SCE),which subsequently undergoes cross-coupling with thein situgenerated radical anions to yield the defluoroborylation products [44].
While in Yang’s protocol (Scheme 13),NHC-BH3is directly oxidized by excited Ir(Ⅲ) species followed by deprotonation to generate NHC-boryl radical.The following addition to CF3-substituted styrene gives anα-trifluoromethyl radical that then undergoes single electron reduction by Ir(Ⅱ) to form a carbanion intermediate.Finally,β-fluoride elimination affords the correspondinggemdifluoroallylboranes.In contrast,NHC-boryl radical is generated by HAT process in Wu’s procedure.The excited photocatalyst oxidizes a thiol catalyst to produce a thiyl radical,which then abstracts a hydrogen atom from NHC-BH3to generate NHC-boryl radical [45].The ensuring transformation including radical addition,SET reduction by the reduced state of the photocatalyst,and fluoride elimination,providesgem-difluoroallylborane products.
4.C–F bond functionalization of trifluoromethyl carbonyl compounds
The carbonyl group is an important unit in organic molecules due to its rich chemistry in further transformations.α,α,α-Trifluorocarbonyl compounds are a class of versatile precursors for the synthesis of di- and monofluorocarbonyl products.Previously,the defluorination of these moieties are exploited using reduction methods with low valent metals as the reducing agents[46]or by electrolysis [47].For example,using Mg as a reducing agent,α,α,α-trifluoroketones are converted to 2,2-difluoroenol silyl ethers,wherein chlorotrimethyl silane (TMSCl) is used to trap the resulting enolates (Scheme 14) [28].The possible mechanism involves a two-electron transfer process,As shown in Scheme 14,the first electron transfer from Mg to ketone gives a ketyl species,which is further reduced to an anionic species by Mg.Afterβfluoride elimination,2-fluoroenol silyl ether is formed as final product.Di-fluoroenol silyl ethers are potentially useful building blocks for various difluoro compounds such asβ-hydroxy ketones 45,α-halodifluoromethyl ketones 46,1,5-dicarbonyl compounds 47.The intramolecular [2+2]cycloaddition affords tetrafluorocyclobutanediols 48.It can also undergo transition metal catalyzed cross coupling reactions to give arylated products 49 and 50.
Samarium(Ⅱ) iodide in conjunction with trimethylamine and water is another single-electron reduction approach used forαdefluorination of esters or amides (Scheme 15) [18].In this defluorination reaction,low temperature (-78 °C) and the addition of Et3N is crucial for controlling the degree of the defluorination process.However,only moderate yield and selectivity could be obtained by this strategy.
Very recently,Wang’s group reported a 4-dimethylamino pyridine-boryl radical promoted sequential C–F bond functionalizations of trifluoromethyl group (Scheme 16) [48].The strategy comprises a controllable two-stage process,each involving a spincenter shift (SCS) pathway for defluorination.In stage A,the reaction starts by the attack of dimethylaminopyridine-BH2•(DMAPBH2•) to the carbonyl oxygen atom of CF3-carbonyl molecules,and the following defluorination occursviaan SCS mechanism,givingα,α-difluorocarbonyl radical intermediates.These intermediates are then reduced by a thiol catalyst or captured by alkenes to afford a wide range of difluorocarbonyl products.In stage A,these difluoro compounds repeat the same process,furnishing diverse monofluoro products.As outlined in Scheme 16,this twostage process shows broad substrate scope and good chemoselectivity.For example,reduction of stages A and B intermediates,wherein RSH was used as the polarity reversal catalyst,can afford di- and monofluoromethyl products selectively,and only minor over-reduction products were observed in the formation of difluoro products.Notably,no trihydrodefluorination product is detected in both cases.Alkenes could be used as the radical trap in both stages A and B,leading to diverse defluorinative coupling products.Interestingly,when two different alkenes were employed in stages A and B,products containing a monofluorinated-tertiary stereogenic center were constructed.
Further DFT calculations revealed that the chemoselectivity is controlled by the declining reactivity of DMAP-BH2•towards the addition to the defluorinated products,which is attributed to the increasing singly occupied molecular orbital (SOMO)/lowest unoccupied molecular orbital (LUMO) gaps between DMAP-BH2•and the substrates.Therefore,once the first fluoride is removed,the resulting carbonyl group becomes less reactive,thus ensuring excellent chemoselectivity during defluorination [49].
5.Conclusion
C–F bond functionalization of CF3-compounds has recently appeared as an efficient method for the synthesis of partially fluorinated molecules.Recent advances on electrochemistry,photoredox catalysis,and radical chemistry of main group elements have rendered more strategies for C–F bond functionalization reactions.In this review,we summarized recent progress on C–F bond functionalization of CF3groups involving radical intermediates as the main transformation pathways.Although the C–F bond in the trifluoromethyl group is extremely inert and the defluorination chemoselectivity is difficult to control,important advancement has been made in selective functionalizations of one or two C–F bonds in CF3groups by different strategies.However,the limitations and challenges are still remained.For example,the sequential functionalization of C–F bonds of trifluoromethylarenes andα,α,αtrifluoroketones with high chemoselectivity has still remained an unsolved challenge.Moreover,the combination of radical chemistry and transition metal catalysis,which is expected to be a powerful tool to construct diversified mono- and difluoro products,has not been well studied.Furthermore,enantioselective transformations of difluoro compounds that are accessed from simple CF3sources has not been achieved yet.The realization of such strategy would of great value to make monofluorinated tertiary stereogenic centers [50]with high enantioselectiity from simple fluorine sources.We can expect that defluorinative functionalization strategies will continue to make important contributions in organic synthesis,medicinal chemistry,and material science.
Declaration of competing interest
The authors report no declarations of interest.
Acknowledgments
We thank the National Natural Science Foundation of China (No.21971226) and the Fundamental Research Funds for the Central Universities (No.WK2060000017) for financial support.
 Chinese Chemical Letters2022年3期
Chinese Chemical Letters2022年3期
- Chinese Chemical Letters的其它文章
- Direct catalytic nitrogen oxide removal using thermal,electrical or solar energy
- Construction and applications of DNA-based nanomaterials in cancer therapy
- Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution
- Nanostructured materials with localized surface plasmon resonance for photocatalysis
- Recent progress of Pd/zeolite as passive NOx adsorber: Adsorption chemistry,structure-performance relationships,challenges and prospects
- Microfluidic methods for cell separation and subsequent analysis