
Neuroprotective effects of Alda-1 mitigate spinal cord injury in mice: involvement of Alda-1-induced ALDH2 activation-mediated suppression of reactive aldehyde mechanisms
2022-06-13 08:32MushfiquddinKhanFeiQiaoPavanKumarTouhidulIslamAvtarSinghJeseongWonInderjitSingh
中国神经再生研究(英文版) 2022年1期
Mushfiquddin Khan , Fei Qiao, Pavan Kumar S.M. Touhidul IslamAvtar K. Singh, , Jeseong Won, Inderjit Singh
Abstract Spinal cord injury (SCI) is associated with high production and excessive accumulation of pathological 4-hydroxy-trans-2-nonenal (4-HNE),a reactive aldehyde, formed by SCI-induced metabolic dysregulation of membrane lipids. Reactive aldehyde load causes redox alteration,neuroinflammation, neurodegeneration, pain-like behaviors, and locomotion deficits. Pharmacological scavenging of reactive aldehydes results in limited improved motor and sensory functions. In this study, we targeted the activity of mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2) to detoxify 4-HNE for accelerated functional recovery and improved pain-like behavior in a male mouse model of contusion SCI. N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide (Alda-1), a selective activator of ALDH2, was used as a therapeutic tool to suppress the 4-HNE load. SCI was induced by an impactor at the T9–10 vertebral level. Injured animals were initially treated with Alda-1 at 2 hours after injury, followed by once-daily treatment with Alda-1 for 30 consecutive days. Locomotor function was evaluated by the Basso Mouse Scale, and pain-like behaviors were assessed by mechanical allodynia and thermal algesia. ALDH2 activity was measured by enzymatic assay. 4-HNE protein adducts and enzyme/protein expression levels were determined by western blot analysis and histology/immunohistochemistry. SCI resulted in a sustained and prolonged overload of 4-HNE, which parallels with the decreased activity of ALDH2 and low functional recovery. Alda-1 treatment of SCI decreased 4-HNE load and enhanced the activity of ALDH2 in both the acute and the chronic phases of SCI. Furthermore, the treatment with Alda-1 reduced neuroinflammation, oxidative stress, and neuronal loss and increased adenosine 5′-triphosphate levels stimulated the neurorepair process and improved locomotor and sensory functions. Conclusively, the results provide evidence that enhancing the ALDH2 activity by Alda-1 treatment of SCI mice suppresses the 4-HNE load that attenuates neuroinflammation and neurodegeneration, promotes the neurorepair process, and improves functional outcomes. Consequently, we suggest that Alda-1 may have therapeutic potential for the treatment of human SCI. Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of MUSC (IACUC-2019-00864) on December 21, 2019.
Key Words: 4-hydroxy-trans-2-nonenal; Alda-1; ALDH2; Basso Mouse Scale score; functional recovery; mitochondrial function;neuroinflammation; neuroprotection; pain; spinal cord injury
Introduction
Spinal cord injury (SCI) causes profound secondary injurymediated neurodegeneration that compromises locomotor,sensory and autonomic functions (Varma et al., 2013; Siddiqui et al., 2015). In spite of high incidence, particularly among young adults, no FDA-approved drug therapy is available for SCI due to limited understanding of druggable targets and cellular injury mechanisms (Ahuja et al., 2017). Although several experimental drugs are in clinical studies, it is critical to identify new and effective drugs for SCI treatment (Wang et al., 2019). Recent advances in medicine indicate that drugs targeting mitochondria-dependent dysfunction are effective in neurodegenerative diseases. Although mitochondrial dysfunction is one of the significant SCI mechanisms (Scholpa and Schnellmann, 2017), the mitochondrial druggable targets’ identity is less clear. Recently mitochondrial enzyme aldehyde dehydrogenase-2 (ALDH2) activation is reported to down regulate inflammation, restore mitochondrial function and improve pain threshold in many animal models (Chen et al., 2014, 2016a; Zhang et al., 2020; McAllister et al.,2021). ALDH2 is the primary enzyme that detoxifies reactive aldehydes, including 4-hydroxy-trans-2-nonenal (4-HNE)(Chen et al., 2014). If not detoxified, 4-HNE forms pathological protein adducts in excess (Gegotek and Skrzydlewska, 2019).
ALDH2 is a NAD(p)+-dependent mitochondrial matrix enzyme. Major substrates (high affinity) of ALDH2 are shortchain aliphatic lipid aldehydes (e.g., 4-HNE, acetaldehyde,malondialdehyde and acrolein). ALDH2 dysfunction has been associated with neurodegeneration, apoptotic cell death,pain and the process of aging (Chen et al., 2014). ALDH2 KO mice had a significantly high 4-HNE load, increased cytokine levels, and persistent behavioral deficits after closed head traumatic brain injury, supporting the anti-inflammatory and antioxidant role of ALDH2 activity in neurotrauma (Knopp et al., 2020). ALDH2 is an established druggable target, and it can be selectively and robustly activated by Alda-1 and inhibited by daidzin (Chen et al., 2014).
4-HNE is a stable and highly reactive lipid aldehyde, and thus it plays a pathological role in neurotraumatic and neurodegenerative disease conditions (Chen et al., 2016a).They can quickly diffuse across the membrane from the site of their origin and accumulate in their adduct form. Therefore,this study is focused on the role of 4-HNE load and its clearance by Alda-1-induced enhanced activity of ALDH2 for the treatment of SCI.
Alda-1 is a potent activator of ALDH2 (Perez-Miller et al.,2010). Pharmacologically, Alda-1 decreases the Km value(Michaelis constant) and increases the V-max (maximum velocity) by binding to ALDH2 near the catalytic site (Perez-Miller et al., 2010; Chen et al., 2014). Alda-1’s half-life is reported 1.67 ± 0.54 hours in rat plasma when 10 mg/kg dose of Alda-1 was administered intravenously (Taneja et al., 2015).The dose of 10 mg/kg body weight has been reported to be effective with significant efficacy in animal studies (Lu et al.,2017; He et al., 2018; Liu et al., 2020).
Based on Alda-1’s efficacy in neurological disorders and nociception, and high safety profile in animal models of CNS diseases, we investigated, for the first time, the therapeutic efficacy of ALDH2 activation using Alda-1 in a mouse model of contusion SCI.
Materials and Methods
Study design
This study’s principal objective was to evaluate the therapeutic potential of Alda-1, an ALDH2 activator, to confer neurovascular protection and aid in functional recovery in a mouse model of contusion SCI. We determined that SCIinduced inhibition of ALDH2 and the sustained overload of reactive aldehydes are reversed by Alda-1, leading to improved pain-like behavior and the accelerated functional recovery. Mice were randomly divided into the sham, control(SCI) and Alda-1 treatment (Alda-1) groups and the number of mice per group was based on our previous studies (Khan et al., 2018) and Basso Mouse Scale (BMS) score-based power analysis. Only male mice were used in this study because traumatic SCI occurs more commonly in males (~80%) than females (~20%) (Chen et al., 2016b). Furthermore, young adult females recover better and faster than young adult males (Swartz et al., 2007). The experimental protocol is described in Figure 1. Experimental groups were blinded, and the biochemical analysis was performed by personnel blinded to groups and samples’ identity.
Reagents
Alda-1 [N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide] (Item# 21555) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Ketamine hydrochloride (ketamine) was obtained from Packager Mylan Institutional LLC, Rockford, IL, USA and xylazine was from Patterson Veterinary, Geeley, CO, USA. All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO,USA) unless stated otherwise.
Animals
Young adult male C57BL/6J mice, aged 10–12 weeks, were purchased from Jackson Laboratory (Bar Harbor, ME, USA).Animals (4/cage) were kept in a helicobacter negative room at ~18–22°C (12-hour dark/light cycle). All animals received humane care in compliance with the Medical University of South Carolina’s (MUSC) guidance and the National Research Council’s humane care criteria. The Institutional Animal Care and Use Committee (IACUC) of MUSC approved animal procedures (IACUC-2019-00864 originally approved on December 21, 2019). Animal number in each group is described in all relevant figure legends.
Mouse model of contusion SCI
A combination of ketamine (90 mg/kg, intraperitoneally[i.p.]) from packager and xylazine (10 mg/kg, i.p.) was used to anesthetize experimental mice. Buprenorphine (0.05–0.1 mg/kg, subcutaneously, once 12 hour) was used as an analgesic, and it was administered pre-emptively and after the surgery to alleviate the pain. The toe was pinched to elicit the withdrawal response. When there is no longer response of pinching, the animal was then placed in a stereotaxic frame(Stoelting Co., Wood Dale, IL, USA). SCI at the T9–10 level was produced on the exposed spinal cord following laminectomy between T9–10. SCI was induced by a computer-controlled impactor device described by Dr. Bilgen (Bilgen, 2005) and used in our laboratory (Chou et al., 2011; Khan et al., 2018).SCI was performed with 1–2 mm tissue deformation and an impact velocity of 1.5 m/s and contusion time of 85 ms.Sham animals had similar procedures, with the exception of the impact. After the completion of the surgical procedures and removal from anesthesia, post-surgical reflexes were monitored. During impact, body temperature was maintained at 37°C. The muscle and skin were closed using nylon suture immediately after injury, and 2% lidocaine jelly was applied to the lesion site. The bladders of all animals were expressed as needed. The body weight and humane endpoints were regularly monitored. The animals were euthanized by decapitation under deep anesthesia to harvest the spinal cord for biochemical and immunohistochemical studies. The spinal cords were stored at –70°C for assays later on if needed.
Alda-1 treatment
Alda-1 was slowly administered at 2 hours after spinal cord contusion. Sham and SCI animals were administered vehicle(20% dimethyl sulfoxide (DMSO)/saline, 100 µL, i.p.). This is the first study on the efficacy of Alda-1 in SCI; therefore, we opted for an early therapeutic time window to establish the efficacy of Alda-1. Delayed treatment time window studies will be determined in our next level studies. In our studies, 10 mg/kg body weight dose of Alda-1 had no effects on physiological parameters (heart rate and body/rectal temperature) when measured at 1 hour after the administration of Alda-1’s preparation (10.0 mg/kg body weight in 20% DMSO/saline,100 µL, i.p.) in mice. Animals in the sham and SCI groups were administered vehicle (20% DMSO/saline, 100 µL, i.p.)
BMS score
The BMS was developed to assess open field locomotion deficits in spinal cord injured mice (Basso et al., 2006). This scoring system is widely used as an indicator of recovery in mouse models of SCI. Later, modifications were added to improve the scale as described (Pajoohesh-Ganji et al., 2010).BMS scoring was performed at the indicated time points, as shown in Figure 1. Two observers, blinded to the identity of groups, independently evaluated the BMS scores.
Video clip of animals’ walking behavior on day 30 after SCI
Animals’ walking behaviors were videotaped using an automated video camera (Canon, VIXIA HF R80, Melville, NY,USA). The entire experiment was performed in an isolated room free from any noise or disturbance.
Evaluation of sensory functions (pain sensitivity)
Pain-like behaviors were assessed by the mechanical allodynia(Khan et al., 2015a) and hyperalgesia (Oghbaei et al., 2020)methods. Improvement in mechanical threshold and thermal withdrawal latency by Alda-1 will indicate improved sensory functions leading to improved pain-like behaviors. The sensory-motor functions were evaluated once a week.
i) Mechanical allodynia: Animals were first acclimatized with a dynamic plantar aesthesiometer (DPA) for about 15 minutes. Dynamic plantar aesthesiometer is an automated version of von Frey hair analysis (Ugo Basile, Italy), and it is used to determine changes in sensation or development of mechanical hyperalgesia (Obata et al., 2004). The animal was placed in an enclosed testing area with a wire mesh on the floor. The dynamic plantar aesthesiometer device was positioned beneath the animal so that the filament directly touched the foot’s surface. The force is increased slowly until it reached 20 g (maximum). The force is recorded at which the foot is withdrawn (paw withdrawal threshold).
ii) Thermal algesia: Mice are gently restrained by placing them in a restrainer, and then the distal 1.0 cm of their tail is dipped into the hot water bath maintained at 49 ± 1.0°C. The latency(time in seconds) between exposure to the hot water and the sudden tail withdrawal is recorded. A 30-second cut-off time was established to minimize the possibility of tissue damage from prolonged heat exposure.
Assay for intraspinal subarachnoid hemorrhage
Mice were perfused with heparinized saline to remove intravascular blood, and a 5 mm segment of cord having the lesion site was homogenized and centrifuged at 16,000 ×gfor 30 minutes. The supernatant containing hemoglobin was left at room temperature for 15 minutes. The absorbance was measured at 540 nm using CLARIOstar microplate reader (BMG LABTECH, Cary, NC, USA). The procedure was validated, and hemoglobin content was determined using known quantities of bovine erythrocyte hemoglobin (Sigma) as described (Lee et al., 2018).
Evans blue extravasation analyses
Blood-spinal cord barrier (BSCB) leakage was determined as described (Cabrera-Aldana et al., 2017) with slight modification. The mice were administered (i.v.) 100 µL of a 5%solution of Evans blue in saline at 4 hours before the 72 hours endpoint. At 72 hours, cardiac perfusion was performed under deep anesthesia induced byketamine/xylazine as described under mouse model of SCI with 200 mL of saline to remove the circulating Evans blue. The spinal cord was isolated and photographed. The spinal cord was homogenized in 750 µL of N,N-dimethylformamide. The homogenized spinal cord was kept at room temperature in the dark for 72 hours and centrifuged at 10,000 ×gfor 25 minutes. The supernatant was analyzed spectrofluorometrically (lex 620 nm, lem 680 nm)by CLARIOstar microplate reader to determine the content of Evans blue.
Measurement of edema (spinal cord water content)
At 72 hours following SCI, mice were euthanized using ketamine (90 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) to assess spinal cord water content (edema) described earlier(Cabrera-Aldana et al., 2017). Fresh spinal cord from was collected, and a segment of 0.5 cm of the spinal cord containing the lesion was weighed. Each sample was dried at 60°C for 72 hours, and the dry weight was recorded. Tissue water content was calculated as: water content (%) = (wet weight – dry weight)/wet weight × 100.
Adenosine 5’-triphosphate assay
Cellular adenosine 5’-triphosphate (ATP) contents in the spinal cords’ injured regions were measured using a firefly luciferase-based ATP assay kit (Bioassay Systems, Hayward,CA, USAl; Cat# EATP-100,) as described (Zhang et al., 2020).Briefly, 20 mg of the tissues were homogenized in 200 µL of cold phosphate-buffered saline, centrifuged at 12,000 ×gfor 5 minutes at 4°C, and the supernatants were collected.The supernatants were equalized based on their protein concentration. A luminescence plate reader measured the ATP level according to the manufacturer’s instructions (CLARIOstar microplate reader).
Western blot analysis
Under deep anesthesia using ketamine (90 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.), the animals at the endpoints were euthanized by decapitation and the spinal cord was collected for biochemical studies. The spinal cords were stored at -80°C for subsequent studies.
Using 5 mm of spinal tissue (~1 mm epicenter; 2 mm caudal and 2 mm rostral from the epicenter) from the injured cord,western analysis was performed as described earlier (Khan et al., 2018). For western blot studies, the following antibodies were used for overnight incubation at 4°C. Intercellular adhesion molecule 1 (ICAM-1; Thermo Fisher Scientific,Waltham, CA, USA, Cat# MA5407, RRID:AB_223596, 1:1000 dilution), 4 hydroxynonenal (4-HNE; Abcam, Cambidge,UK, Cat# ab46545, RRID:AB_722490, 1:1000 dilution),aldehyde dehydrogenase 2 (ALDH2; Abcam, Cat # Ab227021,RRID:AB_2868491, 1:2000 dilution), caspase-3 (pro and cleaved) (Cell Signaling, Danvers, MA, USA, Cat# 9664S,RRID:AB_2070042, 1:1000 dilution), neuronal nuclear protein (NeuN; Abcam, Cat# ab104224, RRID:AB_10711040,1:5000 dilution), ionized calcium-binding adaptor molecule 1 (Iba-1; Abcam, Cat# Ab5076, RRID:AB_2224402, 1:500 dilution), manganese superoxide dismutase (MnSOD; Abcam,Cat# 13533, RRID:AB_300434, 1:2000 dilution), brainderived neurotrophic factor (BDNF; Abcam, Cat# Ab203573,RRID:AB_2631315, 1:200 dilution), neurotrophin-3 (NT-3 Abcam, Cat# Ab263864, RRID:AB_2884942, 1:1000 dilution), cardiolipin synthase 1 (CLS1; Thermo Protein Tech,Waltham, CA, USA, Cat# 14845-1-AP, RRID:AB_2085336,1:1000 dilution), glyceraldehyde-3-phosphate dehydrogenase(GAPDH; Cell Signaling, Cat# 5174, RRID:AB_10622025, 1:5000 dilution), β-actin (Abcam, Cat# Ab8226, RRID:AB_306371,1:10,000 dilution). Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Lab,West Grove, PA, USA, Cat# 111-035-045, RRID:AB_2337938,1:10,000 dilution) was used for 1.5-hour incubation at room temperature. TBS-T (1X) with 2% non-fat dry milk was used to dilute all antibodies. Protein assay kit from Bio-Rad Laboratories (Hercules, CA, USA) was used to determine protein concentration. Tissue homogenates with 20 µg protein were used for western blot analysis and ImageJ software(version 1.47; NIH, Bethesda, MD, USA) was used for the densitometry of protein expression.
Immunohistochemistry and terminal deoxynucleotidyl transferase dUTP nick end labeling assay
At the 30-day endpoint, mice were anesthetized, sacrificed,and perfused first with saline and 4% paraformaldehyde. The spinal cord was decalcified for 48 hours using decalcification buffer, soaked in cryoprotective solution (30% sucrose)for 48 hours, embedded in optimal cutting temperature(O.C.T) compound, and then stored frozen at –80°C. Frozen tissue samples were sectioned transversely (15-µm thick).The sections were stained with an antibody specific to glial fibrillary acidic protein (GFAP; Abcam, Cat# Ab7260, RRID:AB_305808, 1:500 dilution), neuronal nuclear protein(NeuN; Abcam, Cat# Ab177487, RRID:AB_2532109, 1:500 dilution), or myelin basic protein (MBP; Abcam, Cat#Ab62631, RRID:AB_956157, 1:500 dilution) for 1 hour at room temperature. The expression was detected with a secondary antibody conjugated with a fluorescent molecule for 1 hour at room temperature, as described in the manufacturers’instructions and our laboratory (Invitrogen, Carlsbad, CA, USA,Cat# A21206/A32794, 1:1000 dilution) (Choi et al., 2020).
To detect apoptotic cell death, spinal cord sections, prepared as described above, were used for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. According to the manufacturer’s instructions,in situcell death was detected by a kit (Thermo Fisher Scientific, Cat# C10618). Red fluorescent apoptotic cells were manually counted using a BX-60 microscope equipped with a DP70 camera unit (Olympus,Tokyo, Japan).
All digital images were taken using a BX-60 microscope equipped with a DP70 camera unit (Olympus). The acquired images were analyzed by ImageJ software (version 1.47).
Nissl staining
Loss of viable neurons was determined by Nissl (cresyl violet)staining, and thus it reveals the structural features of neurons.The spinal cord sections described above were used to stain with Nissl as previously described (Sakakima et al., 2012). Cells that contained Nissl substance were considered to be viable neurons. Condensed fragmented staining indicates neuronal degeneration. The viable neurons with visible nucleoli were manually counted using a BX-60 microscope equipped with a DP70 camera unit (Olympus).
ALDH2 activity
ALDH2 activity was measured in tissue lysates using an ALDH2 enzymatic activity assay kit from Abcam (Cat# ab115348)according to the manufacturer’s protocol and described by Hua et al. (2018). As described in the western blot analysis section, the frozen spinal cord (5mm segment) was homogenized and centrifuged at 10,000 ×gfor 30 minutes at 4°C. The activity was measured at 25°C. The nicotinamide adenine dinucleotide hydrogen (NADH) production level was determined spectrophotometrically by monitoring the alterations in absorbance intensity at 340 nm every 30 seconds for 5 minutes using CLARIOstar microplate reader.The reaction rates of ALDH2 were expressed as µmol NADH/min/mg protein.
Statistical evaluation
Statistical analysis was calculated as previously (Khan et al.,2015a, b) using software Graph Pad Prism 5.01 (GraphPad, San Diego, CA, USA). All values are expressed as mean ± standard deviation (SD) of n determinations. Group comparisons were performed by one-way analysis of variances (ANOVA) with Tukey’spost hoctest. AP-value less than 0.05 was considered statistically significant.
Results
SCI induces sustained high levels of 4-HNE
Reactive aldehyde load, more prominently of 4-HNE, is recognized as a pathological event in SCI. Western blot(Figure 2A) and its densitometric analysis (Figure 2B) shows a sustained accumulation of significantly high (P< 0.001) levels of 4-HNE adducts at different time points (24, 48, 72 hours,and 30 days) in SCI groups compared with sham-operated (at 30 days) group.
Alda-1 treatment of SCI mice protects against hemorrhage,BSCB leakage, and edema formation by downregulation of pro-inflammatory mediators
BSCB disruption, edema and neurovascular inflammation are critical components of the acute phase of spinal cord contusion injury (Lee et al., 2018). Alda-1 treatment of SCI reduced the severity of contusion injury measured as the blood content in the injured cord (P< 0.01,Figure 3AandB), BSCB disruption (Evans’ blue extravasation,P< 0.01,Figure 3CandD), decreased edema (water content,P< 0.05,Figure 3E) and reduced expression (western blot analysis) of neuroinflammatory mediators ICAM-1 and GFAP (P< 0.01,Figure 3FandG).
Alda-1 treatment of SCI mice enhances the activity of ALDH2 and decreases 4-HNE load in the acute phase (72 hours study) of SCI
4-HNE is one of the most reactive aldehydes, and it is structurally suitable for the formation of protein adducts due to its strong electrophilic nature. Using the two different doses (1 mg/kgvs. 10 mg/kg) of Alda-1, we determined that the effective dose was 10 mg/kg to reduce SCI-induced 4-HNE load (Figure 4AandB). The dose of 10 mg/kg body weight has been effective with significant efficacy in other animal disease models (Lu et al., 2017; He et al., 2018). Therefore, we used a 10 mg/kg dose of Alda-1 in all other experiments reported in this study. The activity of ALDH2 was significantly inhibited in the SCI group compared with the sham group (Figure 4C).Alda-1 treatment of the SCI group significantly increased the ALDH2 activity (Figure 4C). Interestingly, the expression of ALDH2 remained unchanged among the groups (Figure 4DandE).
Alda-1 treatment of SCI mice improves locomotor functions in a 30-day mouse model of SCI
Improvement in the BMS score is the gold standard endpoint determining a therapeutic agent’s efficacy in mice following SCI. On the BMS scores, SCI animals exhibited greater functional loss than the Alda-1-treated group (Figure 5).Statistical analysis showed significantly improved scores in the Alda-1-treated compared with the SCI group from day 3 onward (Figure 5).
Functional recovery was also assessed by the walking behavior on day 30 of SCI and SCI + Alda-1 mice (two mice/group) using video photography. The Alda-1-treated mice had significantly improved walking compared with the SCI animals (Additional Video 1).
Alda-1 treatment of SCI mice improves pain-associated behaviors in a 30-day mouse model of SCI
Both inflammatory and neuropathic pain (caused by a lesion or disease of somatosensory function) are present in most SCI patients (Finnerup, 2013). The ALDH2/Alda-1-dependent mechanism has been reported to mitigate pain by reducing the load of reactive aldehyde in animal models of pain(Zambelli et al., 2014; Li et al., 2018). Using mechanical (paw withdrawal threshold,Figure 6A) and thermal withdrawal latency (Figure 6B), we observed that the pain threshold/latency remained almost unchanged for seven days in both the SCI and the Alda-1-treated groups. From day seven onward, the Alda-1 group had a significantly improved pain threshold that further improved with time (Figure 6). Sham animals had no change in pain sensitivity.
Alda-1 treatment of SCI mice protects against neuronal apoptotic cell death in the chronic phase (30 days study) of SCI
Neuronal cell death is the critical component of a spinal cord contusion injury, and the protection against neuronal cell death is the major objective of SCI therapy. Expectedly, the expression of NeuN was drastically reduced in the SCI group(Figure 7A), which was significantly increased by Alda- 1 treatment (Figure 7AandC). In parallel to NeuN loss in the SCI group, the activity of caspase-3 was increased, as shown by western blot (Figure 7B). In contrast, the Alda-1- treated group had reduced caspase-3 activity (Figure 7BandC). Alda-1 treatment also increased number of neurons measured as the expression of NeuN (Figure 7DandE) and reduced the SCI-induced increased number of TUNEL positive cells (Figure 7FandG), indicating that Alda-1 protects against apoptotic neuronal cell death. The neuroprotective effect of Alda-1 was also supported by Nissl staining showing increased survival of neurons in the Alda-1-treated group (Figure 7HandI). These results suggest a significant neuronal loss in the chronic phase of SCI, and upregulation of ALDH2 activity by Alda-1 may reduce the loss of neuronal cells.
Alda-1 treatment of SCI mice reduces the expression of Iba-1 and GFAP and improves the expression of MnSOD and the level of cellular ATP in the chronic phase (30 days study)of SCI
Inflammation and redox alteration are essential components of SCI, which contributes to neuronal cell death and pain sensitivity, leading to a hindrance to sensory and locomotor functions’ recovery. The expression of activated microglia (Iba-1) (Figure 8AandB) and reactive astrocytes (GFAP) (Figure 8CandD) was significantly increased in the SCI compared to the sham group. Mitochondrial redox, determined as the expression of mitochondria-specific MnSOD, was also compromised considerably in the SCI compared with the sham group. Treatment of the injured animals with Ald-1 decreased the SCI-induced increased expression of Iba-1 and GFAP.The treatment with Alda-1 also improved the SCI-mediated decreased expression of MnSOD (Figure 8EandF) and the reduced levels of ATP (Figure 8G). These results indicate that Alda-1, likely by upregulating ALDH2 activity, is a potent anti-inflammatory and mitochondrial function restoring agent.
Alda-1 treatment of SCI mice increases the expression of neurotrophic factor BDNF, neurotrophin NT3 and myelin structural protein MBP in the chronic phase (30 days study)of SCI
Stimulation of spinal cord repair is essential for optimum functional recovery following SCI (Cheng et al., 1996).Under the oxidative stress environment, as observed in SCI,neurotrophic factors including BDNF and NT3 are reduced,leading to a hindrance to the reparative process. In accordance,western blot analysis showed the reduced expression levels of both BDNF (Figure 9AandB) and NT3 (Figure 9CandD).The expression levels of MBP was also decreased in the SCI group, as indicated by immunohistochemistry (Figure 9EandF). MBP is a critical structural protein that maintains the structure of myelin via interacting with myelin lipids. Reduced MBP levels are associated with demyelination and inhibition of remyelination (Zhou et al., 2019). The treatment with Alda-1 increased the SCI-induced decreased expression levels of BDNF, NT3, and MBP (Figure 9). Taken together, these data indicate that the enhanced activity of ALDH2 and reduced levels of 4-HNE are linked to the stimulation of the expression of neurorepair mediators.
Alda-1 treatment of SCI mice enhances the activity of ALDH2 and decreases 4-HNE load in the chronic phase (30 days study) of SCI
Like in the acute phase, Alda-1 treatment (10 mg/kg but not 1 mg/kg) significantly reduced SCI-induced 4-HNE load in the chronic phase of SCI (Figure 10AandB). While the activity of ALDH2 remained inhibited even at 30-day after the injury, the treatment with Alda-1 enhanced the activity of ALDH2 (Figure 10C). However, like in the acute phase (Figure 4DandE), the expression of ALDH2 had no change among the groups (Figure 10DandE).
Discussion
Enzymatic or non-enzymatic lipid peroxidation (a reaction between polyunsaturated fatty acids and hydrogen peroxide in the presence of free iron) end-product 4-HNE (Ayala et al.,2014) is implicated in several neuroinflammatory diseases,including SCI (Carrico et al., 2009). It is formed in excess and accumulates in CNS traumatic injury conditions such as SCI and traumatic brain injury. Due to an α, β-unsaturated structurebased strong electrophilic nature of 4-HNE, it is highly reactive and forms stable protein adducts (Gegotek and Skrzydlewska,2019). The accumulation of 4-HNE adduct is the consequence of the down-regulated activity of its major metabolizing enzyme ALDH2 in neurodegenerative diseases (Chen et al.,2014). Although 4-HNE is also metabolized by glutathione and aldo keto reductase superfamily enzymes, mitochondrial ALDH2 is the most prominent among them (Ayala et al., 2014).The role of ALDH2 in various neurodegenerative diseases has been recently reviewed by Chen et al. (2016a). Our data inFigure 2provides evidence that 4-HNE load is sustained and prolonged, indicating that 4-HNE adducts accumulation is a pathological event during both the acute and chronic SCI phases. Using a mouse contusive SCI model, we show,for the first time, that suppressing 4-HNE load by Alda-1 via enhancing the ALDH2 activity protects against neurovascular inflammatory injury, mitigates pain-like behaviors, and improves locomotor function.
ALDH2 activity is inhibited by reactive aldehydes. 4-HNE is prominent among such reactive aldehydes. To suppress the 4-HNE load, ALDH2 specific agonist/activator Alda-1 has been used in a number of heart and brain disease conditions (Luo et al., 2014). In this study, Alda-1 treatment of SCI enhanced the ALDH2 activity and reduced the levels of 4-HNE. Unchanged protein expression of ALDH2 among the groups indicates that Alda-1-induced enhanced activity is independent of the ALDH2 expression. No change in the ALDH2 expression levels in a liver ischemia-reperfusion model-treated with Alda-1 (Liu et al., 2020) supports our observation that neither SCI nor Alda-1 regulates ALDH2 protein expression. Alda-1 is reported to increase substrate-enzyme interaction and, thus, the activity (Perez-Miller et al., 2010). Alda-1, via the activation of ALDH2, accelerates the detoxification of only smaller straightchain aliphatic aldehydes such as 4-HNE (Perez-Miller et al.,2010). Interestingly, 4-HNE can diffuse across the biological membranes, and thus it can form adducts with proteins in all cellular compartments (Gegotek and Skrzydlewska, 2019).These observations indicate that Alda-1 directly interacts with the ALDH2 in mitochondria for the up-regulation of ALDH2’s activity, leading to detoxification of 4-HNE and, thus aiding in the amelioration of SCI.
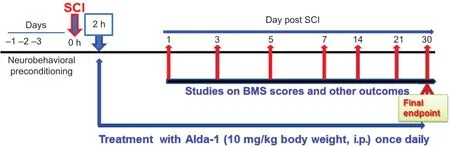
Figure 1|Experimental protocol.
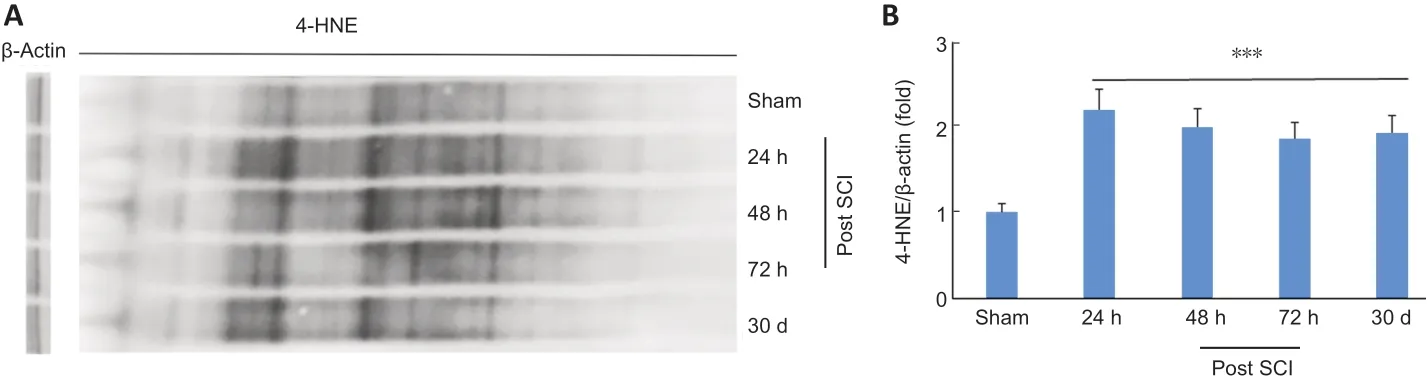
Figure 2|Sustained and prolonged 4-HNE load in a mouse model of SCI.
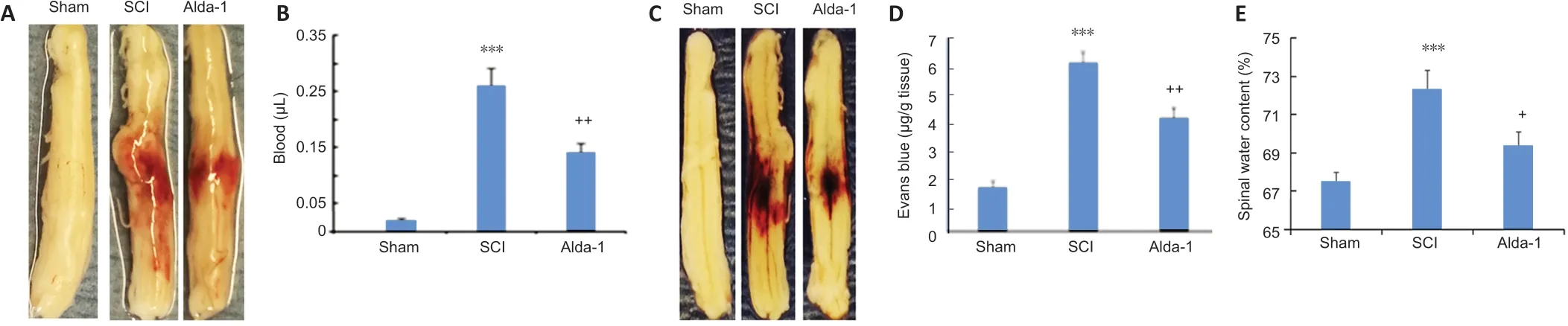
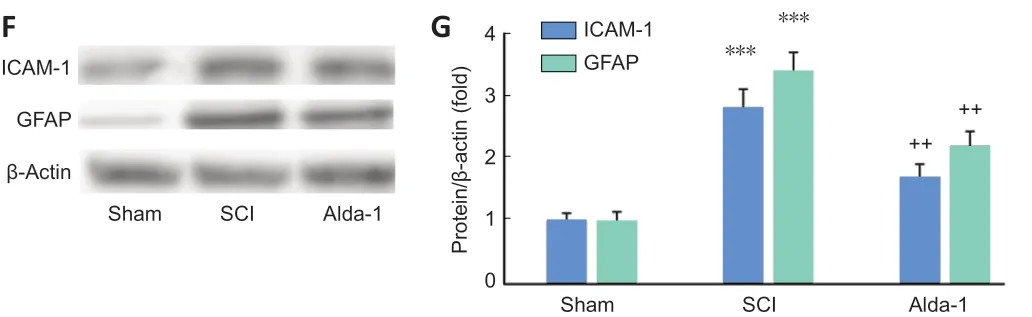
Figure 3|ALDH2 agonist Alda-1 reduces neurovascular dysfunction and inflammation in the acute phase (72 hours) of SCI in wild type mice.
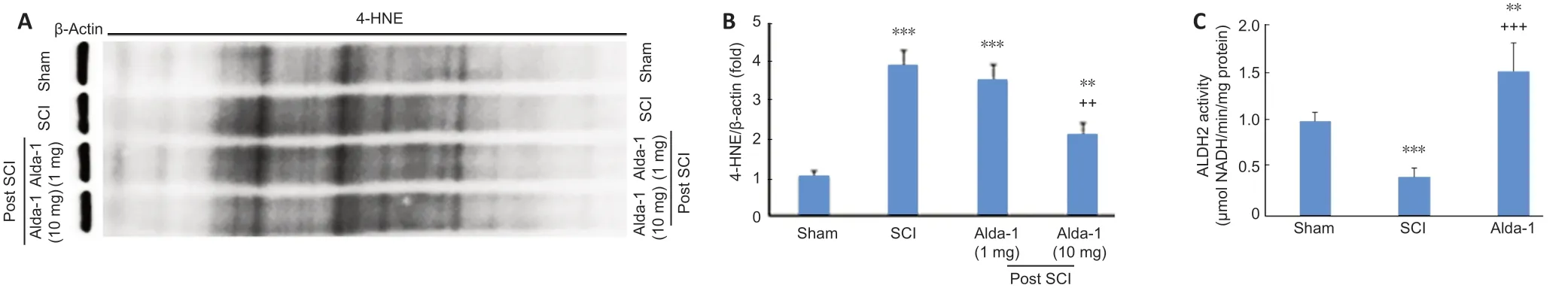

Figure 4|Alda-1 treatment decreases 4-HNE load and enhances the activity of ALDH2 in the acute phase (72 hours) of a mouse model of SCI.

Figure 5|Effect of Alda-1 on locomotor function. Locomotor function was assessed using the BMS scale at indicated days.An observer, blinded to groups, evaluated the BMS.
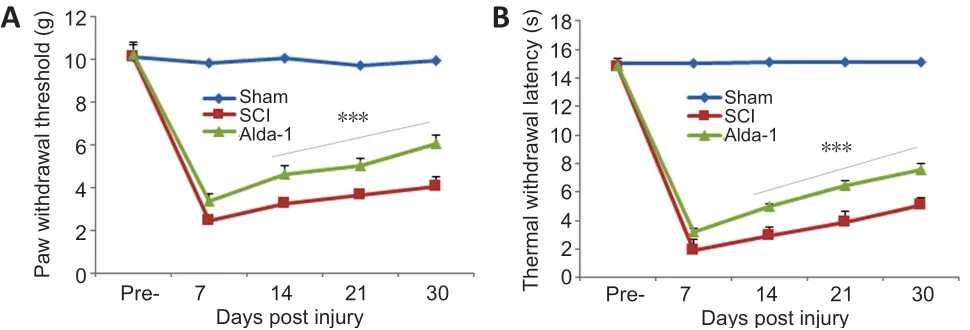
Figure 6|Effect of Alda-1 on pain-like behavior in a mouse model of SCI.
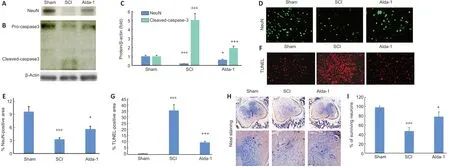
Figure 7|ALDH2 agonist Alda-1 treatment reduces caspase-3 activity, apoptotic cell death, and neuronal loss in the chronic phase (30 days) of a mouse model of SCI.
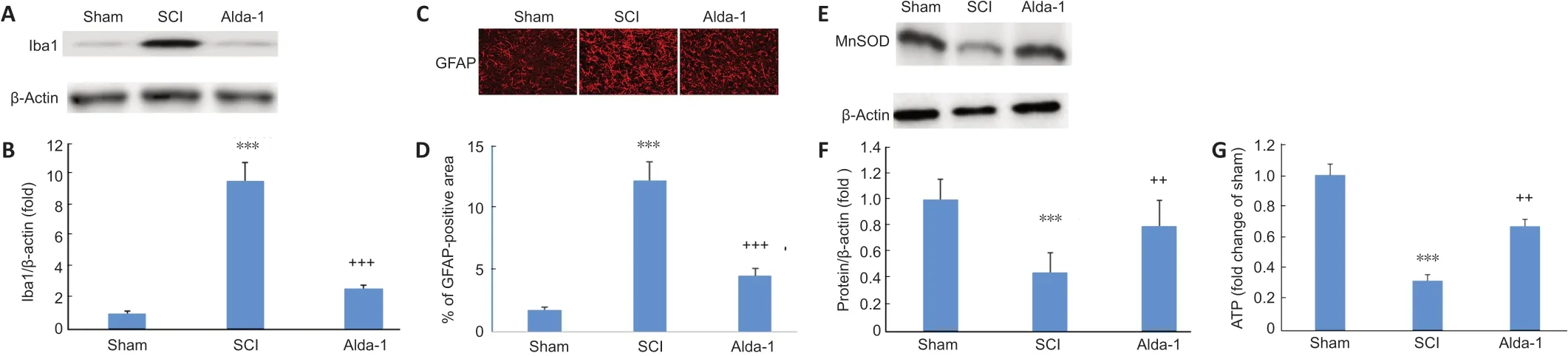
Figure 8|ALDH2 agonist Alda-1 treatment of SCI decreases the activation of Iba-1 and GFAP and normalized mitochondrial oxidative stress measured as the expression of MnSOD in the chronic phase (30 days) of a mouse model of SCI.

Figure 9|ALDH2 agonist Alda-1 treatment of SCI increases the expression of BDNF, NT3, and MBP in the chronic phase (30 days) of a mouse model of SCI.


Figure 10|Alda-1 treatment of SCI decreases 4-HNE load and enhances the activity of ALDH2 in the chronic phase (30 days) of a mouse model of SCI.
In SCI, the primary injury is physical and structural. The initial physical insult resulting from mechanistic crosstalk between several deleterious pathways is followed by secondary injury of oxidative/nitroxidative exacerbations and neuroinflammation(Xiong et al., 2007; Siddiqui et al., 2015; Wang et al., 2019).Therefore, we first investigated whether Alda-1 treatment had neurovascular protective effects on the initial injury mechanisms. Alda-1-induced protection against BSCB leakage and edema and their correlation with the decreased expression of pro-inflammatory mediator ICAM-1 and reactive astrocytes (GFAP). These results indicate an antioxidant/anti-inflammatory property is associated with Alda-1/ALDH2 in SCI. Previous Alda-1 studies, showing protection against endothelial dysfunction and barrier leakage (Lu et al., 2017),corroborate our findings. Our studies and evidence from other studies using Alda-1 (Chen et al., 2008; Perez-Miller et al.,2010; Lu et al., 2017; Zhang et al., 2018) support increasing the ALDH2 activity will ameliorate the acute phase SCI. The alda-1-mediated decrease in 4-HNE load correlates well with the increased activity of ALDH2 in the Alda-1-treated SCI group. These results indicate that neurovascular protection in the Alda-1-treated SCI groups correlates with the increased activity of ALDH2 and decreased load of 4-HNE.
In the chronic phase of SCI, locomotor function deficits, originating from the combined effect of lesion,neurodegeneration, neuroinflammation, and oxidative exacerbations, are the significant consequence of the overall injury. Therefore, the restoration of such functions is always the primary goal of preclinical studies. Evaluation of locomotor functions using the BMS score in mice is the standard gold method (Basso et al., 2006; Qian et al., 2017) to determine the efficacy of an SCI drug. A slow but steady and significant functional recovery, with time, without reaching a plateau,supports Alda-1 therapy’s potential with time. In contrast to the Alda-1 group, the SCI group’s recovery is reaching a plateau. The more significant functional improvement in the Alda-1 group and its correlation with reduced 4-HNE load and the enhanced ALDH2 activity support the beneficial role of Alda-1-induced ALDH2 activation-mediated 4-HNE load suppression strategy. The greater functional recovery in the Alda-1-treated SCI than the SCI group is further supported by a video clip.
In addition to locomotor and sensory function deficits, the pain has a significant impact on SCI patients’ quality of life.The majority of SCI patients suffer from a combination of inflammatory and neuropathic pain (Finnerup, 2013; Hagen and Rekand, 2015). The mechanisms underlying neuropathic pain are multifactorial and complex. Among them, reactive aldehydes, including 4-HNE-mediated mechanisms, are prominent inducing pain-like behaviors (Chen et al., 2014).Our data shows that SCI-induced load of 4-HNE and reduced activity of ALDH2 correlate well with compromised pain behaviors. In contrast, Alda-1 treatment of SCI improved painlike behaviors, indicating that Alda-1-induced ALDH2 activation and 4-HNE load suppression regulate nociception and improve the pain threshold.
After SCI, microglial and astrocyte cell activation correlates with neuroinflammation and indicates that the inhibition of their activation-based drugs may contribute to functional improvements (Pannu et al., 2004; Wang et al., 2019).Increased expression/activation of both microglia (Iba1)and astrocytes (GFAP) after 30-day of SCI indicates SCIinduced inflammation in the chronic SCI. Furthermore,the increased expression of Iba1/GFAP correlated with the decreased expression of mitochondria-specific antioxidant enzyme MnSOD, indicating that neuroinflammation and mitochondrial functions are linked with each other. Because Alda-1-induced neurovascular protective activity is associated with the activation and correction of mitochondrial enzyme ALDH2, increased ATP levels, and 4-HNE load suppression,4-HNE seems to be involved in SCI-induced mitochondrial dysfunction, redox imbalance, and neuroinflammatory events.
The consequence of secondary injury is the accelerated neurodegeneration leading to functional deficits following SCI. 4-HNE-induced neurodegeneration and neuronal loss are well known in a number of neurodegenerative diseases,including animal models of SCI (Xiong et al., 2007; Carrico et al., 2009). Although scavenging 4-HNE is reported to ameliorate CNS trauma, enzymatic detoxification of 4-HNE by enhancing the activity of ALDH2 has not been investigated in SCI. Therefore, ALDH2 activator Alda-1 was a logical choice for 4-HNE load suppression and the neuroprotection following SCI. Remarkable neuronal loss in the SCI group, indicated by significantly reduced NeuN expression as shown by western blot and immunohistochemistry studies, was observed after the four weeks of SCI. In this chronic phase, SCI groups also had increased caspase-3 activity, TUNEL positive, and decreased Nissl positive cells. Furthermore, the expression levels of BDNF, NT3, and MBP were remarkably low in the SCI group. Taken together, these results support that SCI induces apoptotic neuronal loss and neurodegeneration and hinders the stimulation of the neurorepair process. On the other hand, Alda-1 treatment of SCI mitigated SCI-induced neurotoxicity, providing neuroprotection and stimulating the neurorepair process, which correlated well with enhanced activity of ALDH2 and suppressed load of 4-HNE. Taken together, our studies support that enhancing the ALDH2 activity for suppressing 4-HNE load provides neuroprotection,promotes neurorepair process, and aids in functional recovery Several limitations of this study are recognized. First, the data is based on animal experimentsin vivousing wild-type mice,and the cause-and-effect relationship was tested neitherin vitronor in genetically manipulated animals. Second, the study is limited to single-sex (male), and thus, the data may not represent a complete role of ALDH2 activation and the efficacy of Alda-1. We will extend this study to include female animals in next level studies. Third, we investigated only two doses of Alda-1 to determine its effective dose (10 mg/kg).The study is also limited to only one treatment time of Alda-1. We will extend the study to include a delayed treatment time window ranging from 6 hours to 24 hours. Last, the contribution to the SCI of other major reactive aldehydes such as acrolein, formaldehyde, and malondialdehyde, has not been evaluated. However, ALDH2 detoxifies these other aldehydes too. In contrast to beneficial effects, treatment with Alda-1 is also associated with deterioration of renal functions in an ischemia-reperfusion injury animal model due to crystalline nephropathy (Hammad et al., 2018). However, we have not evaluated adverse effects on renal function in this SCI study.
Conclusion
Our data show that Alda-1, a selective agonist of ALDH2,invokes its therapeutic efficacy by targeting the suppression of 4-HNE load in a mouse model of contusion SCI. The study suggests that Alda-1 is a potent antioxidant activitybased drug, and ALDH2 is a druggable target to provide neurovascular protection and aid in functional recovery. Based on our preclinical SCI study’s efficacy and the safety profile in other preclinical studies, Alda-1 seems to be a promising drug to be evaluated in human SCI studies.
Acknowledgments:We thank Ms. Deborah Davis (Department of Pediatrics,MUSC) for her technical help and secretarial assistance. We also acknowledge Dr. Tom Smith from the MUSC Writing Center for his valuable editing of the manuscript.
Author contributions:This study is based on an original idea of MK, JW and IS. MK and SMTI wrote the manuscript and all authors reviewed the manuscript. PK, MK and FQ carried out animal and biochemical studies. MK,AKS, JW, SMTI, IS and PK critically examined the animal and biochemical studies. All authors approved the final manuscript.
Conflicts of interest:The authors declare that they have no conflict of interests.
Financial support:This study was supported by a grant from the State of South Carolina Spinal Cord Injury Research Fund Board, grant No. SCIRF #2017(to MK) and the NIH grant No. R21 NS114433 (to JW and MK). This work was also supported by grants from the U.S. Department of Veterans Affairs, grant Nos. RX002090 (IS) and BX003401 (to AKS). The NIH Grants C06 RR018823 and No C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources also supported the animal work.
Institutional review board statement:All animals received humane care in compliance with the Medical University of South Carolina’s (MUSC)guidance and the National Research Council’s criteria for humane care.Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of MUSC (IACUC-2019-00864) on December 21, 2019.Human data or human tissue has not been used in this study.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional file:
Additional Video 1: Effect of Alda-1 on functional improvement on day 30 after spinal cord injury. Animals’ locomotor/walking behaviors were videotaped using an automated video camera. Two animals from spinal cord injury (white label) and another two from the Alda-1 group (green label) were allowed to move freely in a 2.0 × 2.0 feet box. Alda-1: N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide.

- 中国神经再生研究(英文版)的其它文章
- Genes for RNA-binding proteins involved in neuralspecific functions and diseases are downregulated in Rubinstein-Taybi iNeurons
- Research advances on how metformin improves memory impairment in “chemobrain”
- Dendritic spine density changes and homeostatic synaptic scaling: a meta-analysis of animal studies
- Optogenetic activation of intracellular signaling based on light-inducible protein-protein homo-interactions
- Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease
- Growth differentiation factor 5: a neurotrophic factor with neuroprotective potential in Parkinson’s disease