
Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease
2022-06-13 08:29MercedesHernndezSapinsEdwinRezaZaldvarAnarquezAguirreUlisesmezPinedoJorgeMatiasGuiuRicardoCevallosJuanMateosazctornchezGonzlezAlejandroCanalesAguirre
中国神经再生研究(英文版) 2022年1期
Mercedes A. Hernández-Sapiéns, Edwin E. Reza-Zaldívar,Ana L. Márquez-Aguirre, Ulises Gómez-Pinedo, Jorge Matias-Guiu,Ricardo R. Cevallos, Juan C. Mateos-Díaz, Víctor J. Sánchez-González,Alejandro A. Canales-Aguirre,
Abstract The presenilin genes (PSEN1 and PSEN2) are mainly responsible for causing early-onset familial Alzheimer’s disease, harboring ~300 causative mutations, and representing ~90%of all mutations associated with a very aggressive disease form. Presenilin 1 is the catalytic core of the γ-secretase complex that conducts the intramembranous proteolytic excision of multiple transmembrane proteins like the amyloid precursor protein, Notch-1, N- and E-cadherin, LRP, Syndecan, Delta, Jagged, CD44, ErbB4, and Nectin1a. Presenilin 1 plays an essential role in neural progenitor maintenance, neurogenesis, neurite outgrowth, synaptic function, neuronal function, myelination, and plasticity. Therefore, an imbalance caused by mutations in presenilin 1/γ-secretase might cause aberrant signaling, synaptic dysfunction,memory impairment, and increased Aβ42/Aβ40 ratio, contributing to neurodegeneration during the initial stages of Alzheimer’s disease pathogenesis. This review focuses on the neuronal differentiation dysregulation mediated by PSEN1 mutations in Alzheimer’s disease. Furthermore, we emphasize the importance of Alzheimer’s disease-induced pluripotent stem cells models in analyzing PSEN1 mutations implication over the early stages of the Alzheimer’s disease pathogenesis throughout neuronal differentiation impairment.
Key Words: familial Alzheimer’s disease; familial Alzheimer’s disease-induced pluripotent stem cells models; induced pluripotent stem cells; neurogenesis; neuronal differentiation;Notch; presenilin 1; PSEN1 mutations; γ-secretase complex
Physiological Role of Presenilins
Presenilins (PS) are a family of highly conserved multipass transmembrane proteins located in different cellular structures such as endosomes, lysosomes, nuclear envelope,mitochondria, the trans-Golgi network, and the endoplasmic reticulum (Escamilla-Ayala et al., 2020). PS constitutes the catalytic core of the γ-secretase complex, known better for its proteolytic action on amyloid precursor protein (APP)resulting in amyloid beta-peptide (Aβ) accumulation, one of Alzheimer’s disease (AD) critical events. PS can be classified as γ-secretase-dependent and γ-secretase-independent. Most of the PS functions described up to date are γ-secretasedependent mainly because, as described before, its primary role is to provide the complex’s catalytic core. The γ-secretase complex is a high molecular weight endoprotease that comprises four essential subunits: PS, Pen2, nicastrin, and Aph-1. Among these subunits, PS has shown to mediate most proteolytic events since the secretase activity is dramatically reduced when PS1 is deleted (Oikawa and Walter, 2019). It is noteworthy that some subunits have homologs and isoforms such as PS (PS1 and PS2) and Aph1 (Aph1a and Aph1b),respectively. These variations result in thein vivoassembly of at least six different complexes, though their functional differences are still poorly understood (Yonemura et al., 2016;Stanga et al., 2018).
Many studies have described that multiple γ-secretase complexes have different substrate specificities and have shown that exscind several Type-I transmembrane proteins. The best-studied substrates are APP and Notch for its fundamental implication in AD as well as in cell fate determination and development (Kopan and Ilagan, 2004;Stanga et al., 2018). Nevertheless, numerous other substrates have been identified, including Nectin1a, N- and E-cadherin,ErbB4, LRP, Jagged, CD44, Delta, and Syndecan. These proteins play essential roles in multiple processes, including neural progenitor maintenance, neurogenesis, neurite outgrowth regulation, synaptic function, neuronal function, myelination,and plasticity. Multiple of these processes are disrupted throughout AD pathogenesis, indicating that the complex usually serves as a mediator of diverse signaling pathways(Zoltowska and Berezovska, 2018; Güner and Lichtenthaler,2020).
Search Strategy and Selection Criteria
Literature cited in this review published from 1998 to 2020 was searched on databases including Web of Science,PubMed and Google Scholar. The keywords used were:“PSEN1 mutations”, “Alzheimer’s Disease and PSEN1”,“PSEN1 and neuronal differentiation” “PSEN1 mutations and neurogenesis”, “Familiar Alzheimer’s Disease-induced pluripotent stem cell models”, and “γ-secretase complex and neurogenesis”.
Role of Presenilin 1 on Neural Differentiation
ThePS1protein is ~45–50 kDa in size and is widely expressed in various tissues, comprehending the brain, predominantly in the cell body and dendrites of neurons (Uhlén et al., 2015).Beyond its roles in the APP processing, evidence fromin vivoresearch supports PS1 function during early neurogenesis,with pivotal functions in the proliferation and maintenance of neural progenitor cells (NPC), the appropriate migration of neurons in the cerebral cortex, and the spatiotemporal control of neuronal differentiation and new-born neuron survival capacity (for a detailed review see (Lazarov and Marr, 2010; Bonds et al., 2015). HomozygousPS1-nullmice embryos result in perinatal lethality, displaying severe skeletal deformities, intracranial hemorrhages, and cerebral cavitation associated with massive neuronal loss (Donoviel et al., 1999).Additionally, several studies inPSEN1cKO mice reported decreasing proliferation rates and enhanced differentiation capacity of NPCs (Bonds et al., 2015).
Using selected information where the implication of PS1 in the CNS is evidenced, we created a predicted protein-protein interaction network map with the software STRING (Szklarczyk et al., 2019). The predicted interactome network (Figure 1)emphasizes the implication of PS1 in NPC’s self-renewal and differentiation capacity, highlighting the biological processes of neuron generation in green color, with 17 members interacting and a false discovery rate (FDR) of 3.77E-12 (0.70 of confidence). The dark-blue color shows the canonical WNT signaling with five-member and an FDR of 2.06E-6. In yellow color, Notch signaling is shown with eight members, and an FDR of 2.24E-10. In pink color, the canonical WNT signaling regulation is shown with five members and an FDR of 6.01E-15. The neuronal differentiation is shown in red color with 15 members and an FDR of 5.03E-12. Finally, in light-blue color,the primary molecular function is the protein binding with 19 members and 9.96E-15 FDR.
Here, the best-known biological functions of PS1 are associated with the Notch signaling and neuronal differentiation regulation. Notch proteins are large signaling receptors involved in cell-fate determination and patterning during development. In the adult, Notch activity also is implicated in neural stem cell maintenance, neural differentiation, and regulating neurite outgrowth. Studies using knockouts and γ-secretase inhibitors established a relationship between Notch and PS, observing that the loss of PS activity resembled losses in Notch1 function. Notch signaling can be activated by PS1 throughout the Notch1 intracellular domain delivery and downstream signaling initiation (Bonds et al., 2015; Marathe and Alberi, 2015). Handler et al. (2000) suggested that Notch signaling impairment byPSEN1inhibition results from the downregulated expression of Notch1 downstream target geneHes5. The Notch1 signaling impairment andPSEN1inhibition promote a massive premature neuronal differentiation resulting in a partial depletion of the NPC pools (Handler et al., 2000). Interestingly, reports have shown that Notch signaling also regulates dendrite development in new-born neurons. The conditional knockout of Notch1 in adult-born neurons reduced dendritic branching, while its overexpression significantly increased the dendrite arborization but inhibits neurite outgrowth (Bonds et al., 2015; Ding et al., 2016).
The PS1-induced Notch signaling pathway indirectly mediates the CREB-target genes expression (brain-derived neurotrophic factor and c-fos included), implicated in neuronal survival and long-lasting synaptic changes. This indirect activation appears to be related to the putative CREB-binding protein (CBP)promoter containing a binding site for the Notch-downstream transcription factor CBF1/RBP-Jk (Saura et al., 2004). Besides,Francis et al. (2006) reported a direct PS1-mediated CREB pathway activation that involves the P38 and p42/p44 MAPK and PI3-kinase pathways. In immature neurons, the signaling mediated by CREB is required for initial neuronal dendritic branching by directing dendrite formation and elongation(Bustos et al., 2017), as well to the response to neurotrophic factors promoting neural survival pathways that involve AMP,PKA, MAPK, and genes such as Bcl2 and Mcl1 (Walton and Dragunow, 2000; Landeira et al., 2016). Peculiarly, the PSmediated disassembly of N-cadherin (CDH2) and E-cadherin(CDH1) also affects CREB signaling. A report showed that N-cadherin CTF bind to CREB-binding protein, promoting its degradation, which in turn, downregulates CREB transcription(Marambaud et al., 2002).
E-cadherin and N-cadherin are the best-known cadherins that mediate cell adhesion, with critical roles in the normal development and maintenance of cell-cell contacts. Cadherins bind to β-catenin to adhesion stabilization through the actin cytoskeleton (Marambaud et al., 2002). Reports showed that PS1 might have dual activity regard to cell-cell adhesion:(i) under cell-cell adhesion conditions, PS1 promotes the cadherin-catenin complex anchors to the cytoskeleton,promoting Ca2+-dependent cell aggregation (Baki et al., 2001),(ii) conversely when cell dissociation is needed, PS1 promotes adherents junctions disassembling (Marambaud et al.,2002). The PS1-dependent E-cadherin processing results in a remarkably cytosolic increase of the complex E-cad/CTF2 and soluble β-catenin, a Wnt signaling pathway decisive regulator(Parisiadou et al., 2004; Li et al., 2016). However, there are contradictory data related to PS1 implication in the β-catenin regulation. For example, Killick et al. (2001) reported that PS1 antagonizes the cytoplasmatic and nuclear β-catenin as a response to Wnt1 or Dvl; consequently, there is negative regulation of Wnt signaling-dependent transcriptional activation (Killick et al., 2001). In the absence of PS1, the β-catenin pools are significantly higher, which probably could be related to the hyperproliferative phenotype found inPSEN1null fibroblast (Xia et al., 2001). In contrast, Uemura et al.(2003) suggested that PS1 exert a positive effect in β-catenin nuclear translocation regulation with a subsequent increase of cyclin D transcription, which in turn preceded SH-SY5Y cell differentiation through the β-catenin/TCF/LEF-1 pathway(Uemura et al., 2003; Bonds et al., 2015). Furthermore,another PS1 interacting protein is glycogen synthase kinase 3β(GSK3β), primarily characterized by Wnt signaling involvement.It has been shown that GSK3β phosphorylates PS1 on serine residue 397, regulating its stabilization. Besides, GSK3β influence PS1 ability to associate with β-catenin, improving the stability of the Complex (Duggan and McCarthy, 2016).
During neurogenesis, one of PS’s most relevant functions is the proper migration from neuronal cells’ birthplace to a final location for integration (Wang et al., 2017). Mice with a nullPSEN1die prematurely and display multiple abnormalities,including impairments in cell migration during neocortex development (Handler et al., 2000; Louvi et al., 2004;Buchsbaum and Cappello, 2019). Louvi et al. (2004) reported that loss of PS1 function appears to have a strong link with neuronal migration impairments, compromising radial, and tangential migration. Interestingly, the failure to migrate to an appropriate cortical position in thePSEN1cKO mice correlates with the radial glia reduction and Cajal-Retzius neuron survival(Louvi et al., 2004; Barber and Pierani, 2016).
During the neuronal differentiation, PS1 associates with cytoskeleton components, including MAPT and filamin,regulating neurite outgrowth and stabilization (Zhang et al., 1998a; Pigino et al., 2001). It is unclear how PS1 might mediate this process. Additionally, despite the critical role of presenilins in brain development, their effect during adult neurogenesis remains unclear. In this line, Gadadhar et al.(2011) reported that in neurosphere cultures, downregulation of PS1 improves differentiation without altering their multipotentiality and decreases NPCs’ proliferation. PS1 depletion also downregulates β-catenin and EGFR, implicated in the neural stem cells self-renewal and proliferation regulation (Gadadhar et al., 2011). Also, Bonds et al. (2015)reported that PS1 downregulation reduces p-β-catenin and Notch intracellular cleavage fragments, promoting the cell cycle exit and differentiation. As the expression of β-catenin in NPC is decreased by PS1 downregulation, it may promote a reduction of proliferation-inducing genes (Bonds et al.,2015). In contrast, Dhaliwal et al. (2018) reported thatPSEN1ablation made no changes in maintenance, proliferation,and NPC differentiation. Interestingly, the retroviral-labeled presenilin-null adult neurons show typical electrophysiological properties (Dhaliwal et al., 2018).
To date, there are 149 different substrates identified for the γ-secretase complex. It is unknown if these proteins processing induces aberrant cleavages or generates toxic products, such as Aβ. However, the substrates’ normal biological functions involved in diverse signaling pathways are affected by the γ-secretase complex-mediated processing. Consequently,impaired signaling contributing to neurodegeneration during AD pathogenesis could result from a disruption in the γ-secretase complex (Güner and Lichtenthaler, 2020).
In summary, the reduction of PS1 levels in NPC favors a decreased proliferation rate and enhanced NPC differentiation.Simultaneously, it results in a dendritic branching and spine density reduction and decreased survival rate in newly generated neuronal cells. The predicted interactome network emphasizes the implication of PS1 in NPC self-renewal and differentiation capacity. Hence, the discovery of possible signaling pathways linked to familial Alzheimer’s disease (FAD),and in turn, a possible therapeutic strategy design could be improved by using these types of interactome analyses.
Implication of PSEN1 Mutations in Neural Differentiation
AD is a chronic neurodegenerative disorder characterized by an acute accumulation of neurofibrillary tangles and amyloid plaques in specific brain regions, such as the hippocampus and the cortex, leading to a progressive loss of neurons.Amyloid plaques are insoluble extracellular aggregations of Aβ protein, while intracellularly paired helical filaments composed of hyperphosphorylated tau (an abnormal microtubuleassociated protein) accumulate in the form of neurofibrillary tangles. The Aβ protein is released after the sequential β- and γ-secretase-mediated proteolytic processing of APP (DeTure and Dickson, 2019).
It is well known that most of the AD cases correspond to the sporadic variant of the disorder; however, there is a genetic predisposition variant that involves three different genes originating the disorder. These AD-related genes arePSEN1and2, and APP (encoding for PS1, PS2, and APP proteins respectively) from which mutations in PSEN genes exhibit high penetrance causing the most FAD cases. The most commonly mutated gene and mostly associated with a severe form of the disease isPSEN1, with approximately 319 mutations reported in the Alzforum database (https://www.alzforum.org/mutations/psen-1) (Lanoiselée et al., 2017; Kabir et al.,2020).
The FAD-linked mutations inPSENgenes have been associated with the proteolytic processing of APP because of the γ-secretase-complex implication over the generation of Aβ peptide residues (37 to 49), increasing the self-aggregation Aβ42-residue type abundance, leading to nucleation,oligomerization, and neuropathogenicity, establishing a critical role for PS in AD pathogenesis (de Leeuw and Tackenberg,2019).
There is growing evidence regarding PS1 and APP implication in different physiological processes in neural stem cells and NPC, including differentiation, proliferation, and survival(Figure 2). Consequently, an impaired function or regulation induced by FAD-linkedPSEN1mutations compromise these processes and cause aberrant signaling, synaptic dysfunction, memory impairment, increased Aβ42/Aβ40ratio,aberrant control of neuronal differentiation, contributing to neurodegeneration, mostly in the frontotemporal region(Lazarov and Marr, 2010; Dhaliwal et al., 2018).
It has been suggested thatPSEN1mutations exert detrimental consequences on development because some of them cause very-early-onset AD (before de age of 30 when most aggressive) (DeTure and Dickson, 2019); therefore, many reports have emerged trying to elucidate the roles of PSEN1 mutations over neuronal differentiation (Table 1). Many of them have determined that FAD-linked mutations are mostly associated with dysfunctional mechanisms in the γ-secretase complex (Xia et al., 2015; Watanabe and Shen, 2017).
Furthermore, in neurogenesis, this loss of function causes an aberrant ErbB-4 and E-cadherin-mediated signaling and the downregulation of Notch signaling (Handler et al., 2000;Demars et al., 2010).
Some studies have shown thatPSEN1P117L mutation triggers a decreased neurogenesis by impairing the NPC survival in the dentate gyrus (Wen et al., 2004). In contrast, overexpression of wild typePSEN1in the dentate gyrus did not affect but contributed to neurogenesis (Wen et al., 2002). This finding suggests that this mutation promotes enhanced apoptosis,reduced survival of new neurons, or a neuron-glia switch during fate specification. Similarly, Eder-Colli et al. (2009)reported that thein vitroneurogenesis of murine embryonic NPC was impaired by the FAD-linkedPSEN1P117L, whereas new neurons had enhanced neuritic outgrowth (Eder-Colli et al., 2009).
In comparison to wild-type mice, knock-in mice harboringPSEN1M146V mutation showed an associative learning impairment, consistent with a dentate gyrus neurogenesis reduction. Suggesting that memory deficits related to AD may be due to neurogenesis impairment (Wang et al., 2004).
Veeraraghavalu et al. (2010) demonstrated that in transgenic mice harboringPSEN1ΔE9 mutation, the Notch signaling is altered through the endogenous transcriptional activity of Notch/CBF1 and by the reduction of transcripts encoding Notch-target genes in NPC from the SVZ. Moreover, they showed thatin vitroSVZ NPC cultures derived from mice harboringPSEN1ΔE9 mutation presented a limited capacity for self-renewal and premature differentiation into neurons(Veeraraghavalu et al., 2010). Otherwise, Veeraraghavalu and Sisodia (2013) reported non-transgenic mouse adult hippocampal NPC carrying eitherPSEN1ΔE9 or M146L mutation and showed no dissimilarities in their capacity to differentiate and proliferate compared to wild type. These suggest that impairments in the proliferation and neuronal differentiation capacity inPSEN1mutant mice are due to the expression of thePSEN1mutation by different cells rather than NPC, but residing in the same neurogenic cell niche(Veeraraghavalu and Sisodia, 2013).

Figure 1|Interactome of polypeptides linked to PS1 concerning NPC proliferation and differentiation.
Besides, Demars et al. (2010), in a study using APPswe/PS1PSEN1ΔE9 animal model, suggested that FAD mutations induce early and severely NPC intracellular changes along with altered neurogenic microenvironment in both the hippocampus and SVZ, leading to impaired neurogenesis.They also proposed that these impairments modify the hippocampal and olfactory function, contributing to enhancing the neuronal vulnerability in AD (Demars et al., 2010).
Previously it was suggested thatPSEN1A246E and C410Y mutations affect GSK3β activity (Weihl et al., 1999). Moreover,the GSK3β suppression leads to an enhanced stabilization of β-catenin and TCF/LEF activation, which is essential for neural stem cells self-renewal and NPC proliferation. Hence,the dysregulated activity of GSK3β underlies alterations in tau phosphorylation, neuronal differentiation, neuronal survival,and activation of transcription of neurogenesis-related target genes and proteins (Otto et al., 2016).
Chevallier et al. (2005) reported that mice carrying the PSEN1 A246E mutation showed an increase in β-catenin levels and stimulated hippocampal NPC proliferation without survival or differentiation capacity repercussion, suggesting that this mutation influences NPC cell growth by atypical β-catenin signaling.
Similarly, another study showed thatPSEN1M146L mutation inhibits Wnt signaling by increasing β-catenin phosphorylation and degradationin vitroandin vivo(Kawamura et al., 2001).Therefore, a downregulated β-catenin signaling increases neuronal vulnerability to apoptosis and specific developmental brain defects, whereas restoration of regular β-catenin activity leads to NPC’s pool expansion and neurogenesis restoration(Zhang et al., 2011).
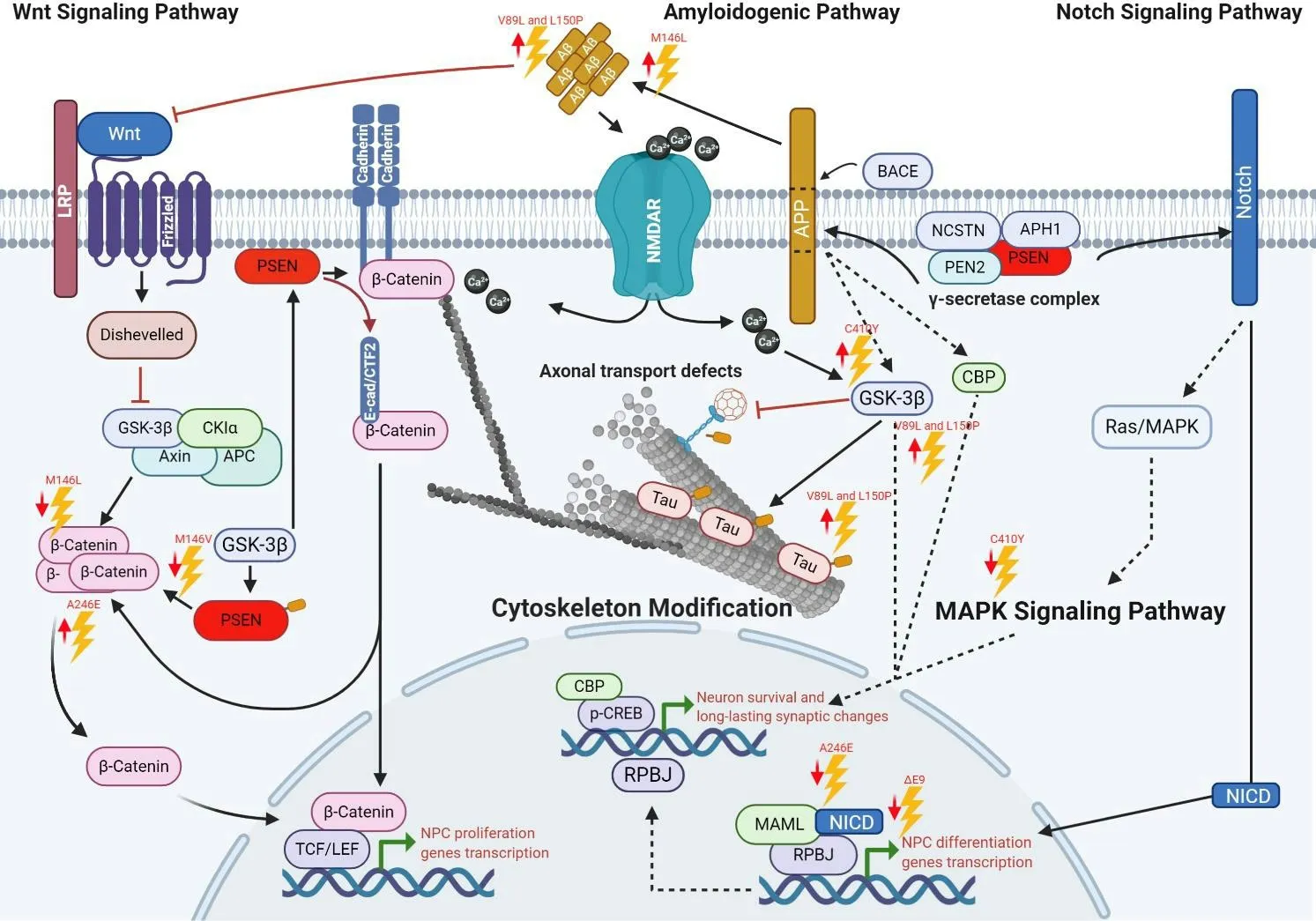
Figure 2|Major pathways disturbed by PSEN1 mutations.
Moreover, Wnt signaling’s loss may induce susceptivity to Aβ-mediated apoptosis (De Ferrari et al., 2003). Thus,PSEN1M146V, C410Y, and I143T mutations promote apoptosis through β-catenin nuclear accumulation and the early-onset AD (Zhang et al., 1998b). In contrast,PSEN1P264L mutant mice did not display any massive neuronal loss, indicating that not allPSEN1-linked mutations lead to neurodegeneration(Siman et al., 2000).
DespitePSEN1implication in several cellular processes, the current transgenic models fail to recapitulate early postnatally events in determined cortical areas and NPC’s pools (Lazarov and Marr, 2010). Additionally, the limited access to ADaffected human tissue made it intricate to study early molecular events during AD pathogenesis.
Modeling Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells
Several research groups reported the generation of human neurons from induced pluripotent stem cells (iPSCs) obtained from FAD patients that share the same genetic background(Yagi et al., 2011). Accordingly, several studies demonstrated that iPSC-derived neurons from AD patients exhibit many AD pathology traits. These studies support the implementation of iPSCs as a platform for a new generation ofin vitroAD models focused on the study of molecular mechanisms,novel biomarkers identification, and novel drugs evaluation(D’Avanzo et al., 2015; Raja et al., 2016; Hernández-Sapiéns et al., 2020; Li et al., 2020).
For example, a remarkable report by Sproul et al. (2014)studied for the first time NPCs derived from FAD-iPSC carrying thePSEN1A246E or M146L mutations. They demonstrated the impact of these mutations in the Aβ42/Aβ40ratios compared with control cells. They concluded that high Aβ42:Aβ40ratios affect NPC’s differentiation capacity andsurvival (Sproul et al., 2014). Similarly, Yang et al. (2017),using the same cell system, showed that thePSEN1mutations induce premature differentiation of NPC, possibly through the upregulation of genes related to neuron development,neuron projection, and neuron maturation. Furthermore,they observed a downregulation in genes mainly associated with cell cycle, which could explain the diminished NPC selfrenewal and elevated apoptosis. Interestingly, they also found a significant reduction in NICD levels, consistent with a study by Borghese et al. (2010), who demonstrated that inhibiting Notch signaling results in neuronal differentiation enhancement (Borghese et al., 2010; Yang et al., 2017).

Table 1 |PSEN1 mutations linked to neuronal differentiation in Alzheimer’s disease
Ochalek et al. (2017), using iPSC-derived neurons carryingPSEN1V89L and L150P mutations, observed a highly active GSK3β, indicating a direct role in Tau hyperphosphorylation(Ochalek et al., 2017). Since Wnt regulates NPC self-renewal by inactivating GSK3β and stabilizing β-catenin, it is expected that an imbalance in the signaling pathway contributes to the dysfunction in NPC maintenance and differentiation capacity.
These and other studies demonstrated the importance of iPSC technology for developingin vitrodisease models that allow us to study the earliest molecular events underlying the pathology. The information obtained fromin vitroAD models can provide the basis for identifying new diagnostic biomarkers, new therapeutic strategies, and the development of “precision medicine”. Additionally, the use of patient-specific iPSC-derived neurons represents a breakthrough strategy for studying the effects of FAD mutations in multiple physiological processes, such as NPC proliferation and differentiation.
Conclusions and Future Perspectives
The PSEN1 protein function and its association with familial AD have been widely studied. Their biological roles’ complexity is becoming increasingly evident because of their implicated cellular processes. Several studies attributed a critical role of PS1 in the neurogenesis process, controlling crucial aspects of the NPC self-renewal and differentiation and in new-born neuron generation. Its implication in dendritic morphogenesis and its subsequent stabilization is remarkable.
To date, FAD is associated with approximately 274 mutations linked toPSEN1. Most of them are missense mutations.Interestingly, the effects of PS1 mutation and their contribution to FAD were first associated with a γ-secretase gain of function due to its ability to influence the APP cleavage and modify the Aβ42/Aβ40ratio. Afterward, another hypothesis suggested a function inhibition by β-catenin absence and Notch intracellular domain and their downstream target genes. Therefore, the biochemical impairment of PS1 is critical in FAD development. In addition to Aβ production, PS1 mutation may alter neurogenesis directly and indirectly, such as NPC self-renewal and differentiation impairment through different signaling pathways, contributing considerably to cognitive impairment.
Despite the relevance of AD research in biomedicine, the molecular pathways leading to neurogenesis impairment and neurodegeneration are still unknown. Moreover, many knowledge gaps remain because of the limited access to ADaffected human tissue and the limited reliability of current transgenic andin vitroAD models. The iPSC technology has revolutionized ADin vitromodels to overcome these gaps by maintaining donors’ genetic information. The analysis of neurogenesis using FAD iPSC-derived NPC offers a unique chance to analyze their biology in a pathological adult environment and the cellular mechanisms regulated by the γ-secretase complex. Thus, an examination of regulated intramembrane proteolysis and its subsequent cellular processes in human neural cells carrying FAD-relatedPSENmutations could provide essential clues to a comprehensive understanding of how these mutations cause AD and how to design novel therapeutic strategies.
Acknowledgments:We would like to thank MD Muhamad Festok from Yale University, United States of America for his valuable effort in editing the English language of the manuscript.
Author contributions:All authors equally contributed to literature search,data collection and analysis, manuscript writing, revision, and approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by the Consejo Nacional de Ciencia y Tecnología Scholarship 711893 (to MAH) and 711874 (to EER).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Genes for RNA-binding proteins involved in neuralspecific functions and diseases are downregulated in Rubinstein-Taybi iNeurons
- Research advances on how metformin improves memory impairment in “chemobrain”
- Dendritic spine density changes and homeostatic synaptic scaling: a meta-analysis of animal studies
- Optogenetic activation of intracellular signaling based on light-inducible protein-protein homo-interactions
- Growth differentiation factor 5: a neurotrophic factor with neuroprotective potential in Parkinson’s disease
- The promise of neuroprotection by dietary restriction in glaucoma