
Genes for RNA-binding proteins involved in neuralspecific functions and diseases are downregulated in Rubinstein-Taybi iNeurons
2022-06-13 08:29LidiaLarizzaLucianoCalzariValentinaAlariSilviaRusso
中国神经再生研究(英文版) 2022年1期
Lidia Larizza, Luciano Calzari, Valentina Alari, Silvia Russo
Abstract Taking advantage of the fast-growing knowledge of RNA-binding proteins (RBPs)we review the signature of downregulated genes for RBPs in the transcriptome of induced pluripotent stem cell neurons (iNeurons) modelling the neurodevelopmental Rubinstein Taybi Syndrome (RSTS) caused by mutations in the genes encoding CBP/p300 acetyltransferases. We discuss top and functionally connected downregulated genes sorted to “RNA processing” and “Ribonucleoprotein complex biogenesis” Gene Ontology clusters. The first set of downregulated RBPs includes members of hnRNHP (A1, A2B1, D, G,H2-H1, MAGOHB, PAPBC), core subunits of U small nuclear ribonucleoproteins and Serine-Arginine splicing regulators families, acting in precursor messenger RNA alternative splicing and processing. Consistent with literature findings on reduced transcript levels of serine/arginine repetitive matrix 4 (SRRM4) protein, the main regulator of the neural-specific microexons splicing program upon depletion of Ep300 and Crebbp in mouse neurons,RSTS iNeurons show downregulated genes for proteins impacting this network. We link downregulated genes to neurological disorders including the new HNRNPH1-related intellectual disability syndrome with clinical overlap to RSTS. The set of downregulated genes for Ribosome biogenesis includes several components of ribosomal subunits and nucleolar proteins, such NOP58 and fibrillarin that form complexes with snoRNAs with a central role in guiding post-transcriptional modifications needed for rRNA maturation.These nucleolar proteins are “dual” players as fibrillarin is also required for epigenetic regulation of ribosomal genes and conversely NOP58-associated snoRNA levels are under the control of NOP58 interactor BMAL1, a transcriptional regulator of the circadian rhythm. Additional downregulated genes for “dual specificity” RBPs such as RUVBL1 and METTL1 highlight the links between chromatin and the RBP-ome and the contribution of perturbations in their cross-talk to RSTS. We underline the hub position of CBP/p300 in chromatin regulation, the impact of its defect on neurons’ post-transcriptional regulation of gene expression and the potential use of epidrugs in therapeutics of RBP-caused neurodevelopmental disorders.
Key Words: alternative splicing; CBP/p300; chromatin regulators; downregulated genes;induced pluripotent stem cell-neurons; neurodevelopmental disorders; ribosome biogenesis; RNA-binding proteins; RNASeq; Rubinstein-Taybi
Induced Pluripotent Stem Cell-Neuronal Model of Rubinstein-Taybi: a Tool for Mechanistic Insights into Intellectual Disability
Rubinstein-Taybi syndrome (RSTS. MIM #180849, #613864)is a rare multisystem developmental disorder characterized by moderate to severe intellectual disability often accompanied by behavior disorder (Ajmone et al., 2018),facial dysmorphisms, including microcephaly and prominent beaked nose, small stature, skeletal dysplasia, multiorgan malformations, and cancer predisposition (Hennekam, 2006).It is caused by monoallelic pathogenic variants in eitherCREBBP(MIM #600140) orEP300(MIM #602700) genes encoding the CBP and p300 lysine (K) acetyltransferases(HATs or KATs) (Sheikh and Akhtar, 2019) with a key role in development and adult life of the nervous system (Valor et al.,2011). RSTS belongs to the expanding group of disorders of the epigenetic machinery (Fahrner and Bjornsson, 2019; Larizza and Finelli, 2019) most of which share intellectual disability.To obtain insights into the molecular basis of cognitive impairment, representing the most devastating clinical sign of RSTS, we established thein vitroinduced pluripotent stem cell(iPSC) neuronal model from peripheral blood ofCREBBPandEP300-mutated patients displaying different levels of cognitive defects (Alari et al., 2018a, b, 2019). Morpho-functional characterization of patients’ versus controls’ neuronal cultures appointed altered morphology of differentiating neurons and reduced excitability of post-mitotic neurons as potential RSTS biomarkers (Alari et al., 2018b). We then compared by means of RNASeq the differentially expressed genes marking the transition from iPSC neural progenitors to cortical neurons of a set of RSTS patients versus a set of healthy controls(Calzari et al., 2020). Consistent with the morphological and electrophysiological data, transcriptome analysis of RSTS iPSCderived neurons (iNeurons) revealed a defective and altered neuroprogenitor to neuron transcriptional program. In brief,transcriptional regulation is weaker in RSTS neurons due to a lower number of modulated genes with respect to control neurons and is subverted by improper upregulation of genes involved in neural migration and axonal targeting and by downregulation of RNA and DNA metabolic genes (Calzari et al., 2020). The latter signature of the RSTS transcriptome is compounded by a large array of downregulated genes (DRGs)associated with the macro-categories of RNA processing (GO Term Cluster 7), Ribonucleoprotein complex biogenesis (G4),telomere maintenance (G2), and DNA metabolic processes(G6) as detailed and discussed in Calzari et al. (2020).
We herein overview the sets of RSTS neuron-specific DRGs(Calzari et al., 2020), associated with the complementary GO term clusters of RNA processing and Ribonucleoprotein complex biogenesis, that encode RNA-binding proteins(RBPs) involved in alternative splicing and ribonucleoprotein biogenesis. The medical relevance of several DRGs pertaining to top and extended DRGs lists (Calzari et al.,2020) is underlined by their links to associated neurological/neurodevelopmental disorders. The high number of DRGs specific of RSTS neurons does not consent analytic comments,but their functional coherence allows discussion of the general network in the framework of the molecular and clinical evidences provided by studies on other human and mouse neuronal systems. Notably, our data give a clue to the recognized connection between chromatin modifiers and alternative splicing (Luco et al., 2010, 2011; Li and Fu, 2019).Indeed, the set of RBPs acting in pre-mRNA splicing and processing predicted at a reduced amount inCREBBP/EP300mutated iNeurons is consistent with the impact of CBP/p300 depletion on the regulation of cryptic splice sites (Blazquez et al., 2018; Boehm et al., 2018) and the highly conserved neuron-specific microexons splicing program (Gonatopoulos-Pournatzis et al., 2018; Lopez Soto et al., 2019). Functional links between the epitranscriptome and the RBP-ome are also highlighted by DRGs for RBPs involved in ribonucloprotein biogenesis as some of these proteins perform as “dual”players. The overall signature of DRGs in RSTS iNeurons informs on the negative repercussion of defective CBP/p300 on multiple RBPs which cumulative dysfunction contributes to cognitive impairment.
Search Strategy and Selection Criteria
We searched the literature published in the period 2000-2020 using the terms in the Title, Abstract and Keywords on PubMed and retrieved further articles by Pub Med related functions and citation tracking. Due to the fast-growing field of RNA binding proteins we prioritized contemporary literature with the exception of milestones papers. OMIM, Gene Cards,Human Gene Mutation Database, Chromatin databases were relevant tools in our study. The major inclusion criteria were literature on the link between chromatin dysregulation and altered RNA binding proteins assembly and their impact on neuronal functions and neurodevelopmental disorders.
RNA-Binding Proteins
RNA-binding proteins (RBPs) are a large class of > 2000 proteins, which contain RNA binding domains (RBDs) for the purpose of binding target RNAs via specific sequence motifs(Gerstberger et al., 2014b; Corley et al., 2020). They interact with all known classes of coding and non-coding RNAs making up assemblies referred to as RNA-protein complexes (RNPs)(Gerstberger et al., 2014b). The RNPs regulate gene expression at all stages of the mRNA life cycle, encompassing alternative splicing (AS) and nucleocytoplasmic shuttling, mRNA localization, stability, and translation (Hentze et al., 2018). In the nervous system RNPs regulate the spatio-temporal gene expression to adjust neuronal functions according to the ever-changing environment, and to insure that RBP-select mRNAs are transported to subcellular compartments such as dendrites and axons for localized translation (Thelen and Kye, 2019). AS has emerged as the fundamental mechanism for providing the heterogeneous neurons with the unique repertoire of protein isoforms needed for development of neural cell type-specific properties, synapse specification, and establishment of functional networks (Furlanis and Scheiffele,2018). It is thus not surprising that mis-regulation of RBPs leads to development of an increasingly recognized number of disorders mostly of neurologic, muscular, and sensory origin (Lukong et al., 2008). About 200 RBPs are recorded in the OMIM database (https://www.ncbi.nlm.nih.gov/omim) as being associated with human diseases, attesting to their key role in post-transcriptional regulation of gene expression.
Many excellent reviews have covered the classification of RBPs, based on the structure and function of their RBDs,binding preferences and target RNAs, high conservation across species, and ubiquitous or tissue-specific expression(Gerstberger et al., 2014a, b; Neelamraju et al., 2015). The technical approaches for studying system-wide protein-RNA interactions and the transcriptomic and computational methods for tackling RBPs interaction networks have been expertly reviewed (Hentze et al., 2018; Sternburg and Karginov, 2020) as well as the software cataloging or predicting RBPs and their targets (Corley et al., 2020).
As most RBPs are multitasking, there are limitations in their assemblage by function. However clustering them by class is biased due to the continuous update of RBPs census(Gerstberger et al., 2014a), as the function of at least onethird of RBPs is unknown and the list of “unconventional”RBPs, lacking discernible RBDs, is growing over time (Hentze et al., 2018). Given that members of different RBPs families act in concert on specific cell processes, we grouped the univocal DRGs of RSTS iNeurons based on their main shared function to underscore weakened gene networks at the root of RSTS neurodevelopmental disorder.
Genes for RNA Binding Proteins Are Downregulated during Differentiation of Rubinstein-Taybi Induced Pluripotent Stem Cell-Neurons
RSTS-univocal top DRGs include about 30 members of diverse RBP families. A similar collapse of the post-transcriptional regulation machinery has been observed in the transcriptome of iNeurons from patients with autism spectrum disorders(DeRosa et al., 2018) and cardiomyocytes from patients with Cornelia de Lange, an intellectual disability syndrome often presenting with autistic traits (Mills et al., 2018). The shared signature of distinct neurodevelopmental disorders may originate by the failure to safeguard the highly dedicated post-transcriptional regulation program during differentiation to neurons and cardiomyocytes when transcription hubs such as the autism and the cohesin genes, forming critical interactions in the 3D genome with the splicing regulation network, are damaged. As regards RSTS, it is predictable that a defect of either CBP or p300 multifunctional transcriptional coactivators, that acetylate a multitude of diverse signaling effectors and enhancer-associated regulators (Weinert et al., 2018) might subvert the RBP-ome profile, especially of proteins involved in RNA AS and processing, a strictly regulated process in neuronal cells (Furlanis and Scheiffele,2018). Furthermore, functional cross-link between chromatin regulation and alternative splicing has been documented in a range of organisms favoring an integrated model of the role of chromatin and histone modifications for alternative splice site selection and other RNA processing events (Luco et al.,2010, 2011). Splicing of very short exons (3–27 nt), a highly conserved program of AS in neural cells, has been revealed to be highly relevant in shaping protein-protein interactions associated with brain-specific functions (Irimia et al., 2014)and has been linked to autism-spectrum and psychiatric disorders by genome-wide CRISPR-Cas9 interrogation of splicing networks (Gonatopoulos-Pournatzis et al., 2018).Remarkably, these screens showed that several genes impact the main SRRM4 splicing regulator of the activity-dependent neuronal microexons, and act at multiple regulatory levels,including RNA processing, chromatin regulation, and protein turnover (Gonatopoulos-Pournatzis and Blencowe, 2020).CBP/p300 are the main chromatin regulators impacting this network, as demonstrated by reduced SRRM4 levels upon their depletion in mouse neuronal cells (Gonatopoulos-Pournatzis et al., 2018).
Our overview is focused on DRGs sorted to the most expanded Go term Clusters “RNA processing” and “Ribonucleoprotein complex biogenesis” with several “multifunctional” DRGs acting in both processes (Calzari et al., 2020). Top DRGs(padj cutoff < 1 × 10–3) and functionally linked, though less significant, DRGs are discussed.
Downregulated RNA Binding Protein Gene Families Involved in Alternative Splicing
Several univocal RSTS DRGs encode members of RBPs families which act in a synergistic/antagonistic way in AS regulation.Some encode components of the spliceosome, a dynamic multi-megaDalton ribonucleoprotein (RNP) complex that executes precursor messenger RNA (pre-mRNA) splicing by recognition of core sequence elements (van der Feltz et al.,2012), while others encode trans-acting regulators which recognize by their RBDs the target RNAs. The spliceosome catalyzes the removal of introns and ligation of exons: splicing choices are largely achieved by cis-acting elements referred to as exonic enhancers and intronic silencers that recruit RBPs for binding (Hentze et al., 2018). The contribution of RBPs to AS is to regulate the access of the core splicing machinery, which is strongly dependent on the cell-specific RBP-ome and the position (relative to the regulated exon) of the RNA binding motif (Furlanis and Scheiffele, 2018). AS shapes the entire life cycle of a neuron from early differentiation to axonal guidance and synapse formation and signaling till maintenance of neural circuit function response to a dynamic environment(Furlanis and Scheiffele, 2018; Lipscombe and Lopez Soto,2019). Over 90% of human pre-mRNAs are subject to AS (Wang et al., 2008) and the splicing decisions are regulated in a cell type- and cell state-dependent manner. In the nervous system the capacity of multi-exon genes to generate a huge number of splice variants to diversify protein isoforms across brain regions and developmental stages (Furlanis and Scheiffele,2018) and to transport selected transcripts within neurons’subcellular compartments for local translation has proven indispensable for development, synaptic plasticity and long term memory (Bramham and Wells, 2007). Indeed, the magnitude of AS has remarkably increased in organisms with more complex nervous systems, such as mammals (Furlanis and Scheiffele, 2018). A specific and highly conserved program for recognizing and splicing microexons has evolved in the mammalian nervous system and its perturbation has been linked to autism and cognitive dysfunction syndromes (Irimia et al., 2014; Gonatopoulos-Pournatzis et al., 2018). AS also produces mRNA variants that are substrates for nonsensemediated mRNA decay, which serves to eliminate aberrant mRNA transcripts with premature termination codons and to finely regulate post-transcriptional regulation (Lykke-Andersen and Jensen, 2015). Missplicing events leading to exon skipping or intron retention (Wang et al., 2008; Das et al., 2019) have severe repercussions on neuronal functions, as indicated by the growing number of diseases of the nervous system characterized by aberrant splicing, either driven by mutations that disrupt splicing directly by interfering with cisacting elements and trans-acting splicing factors or indirectly by sequestering RNPs to visible membraneless nuclear or cytoplasmic compartments with an arrested translation of their scaffold RNA (Lipscombe and Lopez Soto, 2019).Pervasive transcriptome-wide isoform-level deregulation in the human brain has been reported to be associated with autism spectrum disorders, schizophrenia, and bipolar disorders (Gandal et al., 2018).
Additional Table 1outlines the DRGs in RSTS iNeurons encoding RBPs members of heterogeneous nuclear ribonucleoproteins (hnRNPs) family, Small Nuclear Ribonucleoprotein Polypeptides (snRNPs), and Serine and Arginine splicing factors (SRSF), acting in pre-mRNA AS either cooperatively or in competition for overlapping binding sites.
hnRNPs
hnRNPs, referred to as “mRNA clothes” as they bind nascent transcripts produced by RNA polymerase II (heterogeneous nuclear RNA hnRNAs/pre-mRNA) and supervise their maturation into messenger RNAs (mRNA), stabilization,transport and translation, have been intensively investigated for their relative abundance (> 50% of RBPs), multifunctional regulatory roles and dysfunction in neurodegenerative/neurodevelopmental disorders (Chaudhury et al., 2010; Han et al., 2010; Geuens et al., 2016). hnRNPs are ubiquitously expressed and their main function is to control cryptic exon inclusion and inhibit cryptic polyadenylation sites safeguarding the integrity of the transcriptome (Das et al., 2019).
The human genome contains about 40 hnRNP genes comprising distinct subfamilies originating via duplication events, designated alphabetically from hnRNPA1 to hnRNPU. All contain one or more modular RBDs out of the~600 structurally distinct ones which have been described(Gerstberger et al., 2014a; Corley et al., 2020). The most represented RBDs include RNA Recognition Motifs (RRM),quasi-RRM (qRRM), K-Homology RBD and/or RGG boxes consisting of Arg-Gly-Gly repeats with interspersed aromatic amino acids (Phe/Tyr) and Gly-, Arg-, Lys-, Pro-rich auxiliary domains for homologous and heterologous interaction with RNA or proteins (Han et al., 2010). These latter motifs are typical of C-terminus intrinsically disordered regions (IDRs)which, despite their lack of structure, act as RBDs driving the majority of protein-RNA interactions in the cell and are enriched among the hubs in protein interaction networks(Neelamraju et al., 2015; Hentze et al., 2018).
Each hnRNP family member differs in the number and orientation of its subunits whose modularity contributes to the combinatorial nature by which they associate with different RNA targets (Levengood and Tolbert, 2019). Most hnRNPs have a nuclear localization signal (NLS) and are predominantly present in the nucleus during steady-state, appearing at electron microscopy in large nucleosome-like complexes referred to as “ribonucleosomes”, a term pointing to their main function of packaging pre-mRNAs (Chaudhury et al.,2010). Upon post-translational stimulation or recruitment of other hnRNPs they shuttle to the cytosol where often undergo post-translational modifications (acetylation, phosphorylation,methylation, small ubiquitin-like modification) and regulate mRNA localization, stability, and the efficiency with which specific mRNAs are translated (Han et al., 2010; Geuens et al.,2016). Though hnRNPs are clustered in one family because of some commonalities and shared functions, their structural and functional divergence is increasingly recognized making their distinction from RBPs questionable (Han et al., 2010).In the nervous system where highly polarized neurons, with axons as long as one meter, are highly dependent on local translation and a transcriptome of > 2500 mRNAs is present at synaptic regions (Cajigas et al., 2012), hnRNPs are crucial for transporting the stabilized mRNAs one by one along the axonemal cytoskeleton to the subcellular hubs of protein translation. Deregulation of hnRNPs dosage and dynamics due to persistent cellular stress or mutations disrupting neuronal ribostasis and proteostasis is at the root of several neurodegenerative and neurodevelopmental disorders(Nussbacher et al., 2019).
Additional Table 1shows the major hnRNP A1, A2B1, D, G(RBMX), H1/H2, and the minor MAGOHB and PABPC1 family members downregulated in RSTS neurons. Their structural domain organization is shown inFigure 1and summarized with their functions, and their link to constitutional diseases inAdditional Table 2.
hnRNPA1-hnRNPA2B1
A1 is the prototypical and best-known member of the hnRNP family. Composed of 2 RRM domains, close to one another and similar in size, and 1 C-ter IDR with a RGG box, it is structurally similar to A2B1 (2 RRM and 1 C-ter IDR with an RGG box) (Figure 1andAdditional Table 2). The RRMs are the primary RNA binding surface, whereas the IDRs mediate protein-protein interactions. The presence within the G-,Y-rich IDRs of mammalian-specific alternative exons mediates the formation of tyrosine-dependent multi-hnRNP assemblies that globally regulate splicing patterns, highlighting a mechanism that has expanded the AS regulatory capacity of mammalian cells (Gueroussov et al., 2017). On the other hand, due to the repeats of non-polar (Gly) and aromatic amino acids (Phe/Tyr),the IDRs of these hnRNPs are “prion-like domains“ (PrLDs),i.e. protein conformers that self-replicate by templating the folding of soluble proteins with the same amino acid sequence(Harrison and Shorter, 2017; Levengood and Tolbert, 2019;Liu and Shi, 2020). PrLDs enable RBPs to undergo liquid-liquid phase separation, a process underlying the biogenesis of various membraneless organelles, such as Cajal bodies in the nucleus and stress granules in the cytoplasm, which are highly dynamic and assemble and disassemble according to the local environment. Inappropriate transitions due to loss of neuronal homeostasis or mutation render these proteins prone to misfolding and aggregation around the mRNA scaffolds,thus reducing the pool of functional protein, and interfering with its normal function, outlining a common pathway to neurodegeneration (Harrison and Shorter, 2017; Picchiarelli and Dupuis, 2020).
hnRNPA1 and hnRNA1B2 are multitasking proteins with a major role in the regulation of AS and splice-site selection,often promoting exon skipping (Jean-Philippe et al., 2013;Liu and Shi, 2020). They are essential for the maintenance of transcriptome integrity by protecting thousands of endogenous pre-mRNA transcripts with highly complex sequences against mis-splicing (Das et al., 2019). Both have a role in nucleocytoplasmic transport, stability, and degradation of mature mRNA transcripts, mRNA translation, telomere biogenesis, and length maintenance (Han et al., 2010; Shishkin et al., 2019). A1 also participates in microRNA processing. A1 and A2B1 are among the most abundantly expressed proteins in the cells, only rivaling histones, and form “ribonucleosomes”with many other different hnRNPs. In the cytoplasm, they are involved in RNP granules assembly whose packaging in conditions of cellular stress or mutation leads to the arrest of mRNA translation and accelerated fibrillization of the altered protein (Picchiarelli and Dupuis, 2020). Differently from hnRNPA1, hnRNPA2B1 plays a crucial role in mRNA trafficking in neurons and oligodendrocytes and is important for the localization of transcripts containing an A2 response element(Liu and Shi, 2020). It is a reader of N6-methyladenosine (m6A),the most abundant internal modification of mRNA, and binds the nuclear modified transcripts to elicit their alternative splicing and processing (Alarcon et al., 2015). A2B1 has been recognized as an innate sensor of DNA virus infection in the nucleus, able to elicit and amplify type I interferons responses,also promoting m6A modifications (Wang et al., 2019;Additional Table 2).
Whole exome sequencing and linkage analysis detected mutations in the C-ter PrLDs of hnRNPA1 and hnRNPA2 (the shorter hnRNPA2B1 isoform) in families affected by a rare multisystem proteinopathy formerly known as Inclusion Body Myopathy with Paget’s bone disease, frontotemporal dementia, and amyotrophic lateral sclerosis (ALS) (Benatar et al., 2013; Kim et al., 2013). Additional hnRNPA1 and hnRNPA2 mutations were detected in patients with sporadic and familial ALS, though their prevalence in ALS is unknown (Harrison and Shorter, 2017). The pathogenic role of these hnRNPs may go beyond the rare cases of germinal mutations as attested by their widespread colocalization in stress granules with other frequently mutated ALS proteins (Picchiarelli and Dupuis,2020). It has been claimed that RBP homeostasis should be considered globally and that consequences on multiple RBPs could be expected upon mutation of one single member.Based on accumulation and colocalization with other mutated RBPs, both A1 and A2B1 could be involved in several muscular and neurological diseases (Picchiarelli and Dupuis, 2020;Additional Table 2).
hnRNPD, alias AUF1 (AU-rich element binding factor 1)comprises 4 isoforms generated by AS constituted by two non-identical RRM and a C-ter IDR with several RGG motifs which hosts a nonclassic nuclear localization signal(Figure 1). The RRMs serve to form complexes with AUrich elements in the 3′-untranslated regions of many protooncogenes, cytokines, and circadian clock gene mRNAs: as a result, hnRNPD plays a defined role in destabilizing mRNAs,often associated with accelerated mRNA decay (White et al., 2017). Besides binding in a specific manner to singlestranded telomeric DNA (Shishkin et al., 2019), hnRNPD binds chromatin DNA functioning like a transcription factor. It has been recognized as a new player in DNA double-strand break repair via homologous recombination, able to localize at the sites of damage and to act in the DNA-end resection process(Alfano et al., 2019;Additional Table 2). hnRNPD is comprised with its hnRNPDL paralog within the commonly deleted region of the 4q21 microdeletion syndrome (Additional Table 2)characterized by severe psychomotor retardation, marked growth restriction, distinctive facial features and absent or severely delayed speech (Bonnet et al., 2010).
hnRNPG, alias RBMX (RNA Binding Motif Protein X-linked),contains an N-terminal RRM, a large low complexity Ser-Arg-Gly-Pro-rich region, and a C-terminal RBD (Figure 1).The IDR region which self assembles into large particlesin vitrocomprises the nascent transcripts targeting domain,involved in the recognition of an RNA motif exposed by m6A modification (Liu et al., 2017). The hnRNPG protein ubiquitously controls pre-mRNA AS site selection and can either activate or suppress exon inclusion of several neurological disease-associated genes (Wang et al., 2011).Its role in neural development is attested by the association of a C-terminal truncating mutation to a human intellectual disability syndrome (MIM #300238) (Shashi et al., 2015).Besides being involved in DNA damage repair, chromatid cohesion, and transcriptional regulation, hnRNPG is an m6A reader, using its IDR to bind the exposed modified purine rich-motif, contributing to the AS regulation of m6A modified mRNA transcripts (Liu et al., 2017;Additional Table 2).
hnRNPH1 and hnRNPH2, also named hnRNP and hnRNPH’,sharing 96% homology and encoded by theHNRNPH1andHNRNPH2genes, possess 3 non-conserved highly homologous RNA Recognition Motifs (qRRM), one glycine/tyrosine/arginine-rich domain-containing a nuclear localization signal(NLS), and one glycine/tyrosine-rich domain (Figure 1). The best-known function of hnRNPH1/2 is the control of AS:both are repressors of neurogenesis, in part by preventing the expression of the pro-differentiation TRF2-s (Telomere-Repeat Factor 2-short isoform), though H1 appeared to have a stronger splicing effect (Grammatikakis et al., 2016). A role for H1 in the control of human stem cell fate has been highlighted by demonstrating that its reduced expression modifies the mutually exclusive isoforms of T-cell Factor 3 transcription regulator during differentiation of pluripotent human ESCs(Yamazaki et al., 2018) (Additional Table 2). A study reported on the role of hnRNPH sequestered together with other hnRNPs in toxic RNA foci in the spinal cord of amyotrophic lateral sclerosis/frontotemporal dementia patients (Cooper-Knock et al., 2015).
Monoallelic pathogenic variants, believed to act by gainof-function mechanism, were first identified inHNRNPH2by whole exome sequencing on patients with syndromic intellectual disability (Bain et al., 2016). As all the first mutation carriers were females, the new syndrome (MRXSB,MIM #300986), characterized by intellectual disability,dysmorphic features, feeding problems, and hypotonia, was thought to be potentially lethal in males, but males with features consistent with MRXSB were later reported, together with additional females (Harmsen et al., 2019; Jepsen et al.,2019; Peron et al., 2020; Somashekar et al., 2020;Additional Table 2).
The first pathogenic variant in the paralog autosomalHNRNPH1gene was reported in a patient whose phenotype partially overlapped that ofHNRNPH2-caused syndrome but included unique features (Pilch et al., 2018), a finding confirmed by the description of seven newHNNRPH1-mutated patients (Reichert et al., 2020). It is noteworthy that most variants in both genes affect the glycine/tyrosine/arginine-rich domain containing the nuclear localization signal, between amino acids 194 and 220, with the 205–213 stretch highly conserved and required for nuclear transport,suggesting improper nucleocytoplasmic shuttling of the pathogenic variants. The c.616C > T, pArg206Trp variant is shared by 10/12HNRNPH2- and 5/8HRNRPH1- mutated patients reported so far (Figure 2). Further patients need to be characterized to delineate the phenotypic spectrum of this new neurodevelopmental syndrome and assess its common/unique features in comparison with theH2-caused neurodevelopmental disorder. Notably, both disorders share developmental and speech delay, a few dysmorphic features,feeding problems, and skeletal issues with RSTS. However,with the limitation of the small number of patients,HNRNPH1-mutated patients share additionalH1-unique features with RSTS, such as MRI brain anomalies, congenital microcephaly,palate abnormalities, distinctive dysmorphisms such as long prominent nose with hypoplastic alae nasi and low hanging columella, micrognathia and congenital malformations of ocular and urogenital systems (Pilch et al., 2018; Reichert et al., 2020). This clinical resemblance may be accounted for by the finding thatHRNRPH1, unlikeHNRNPH2, is downregulated in RSTS iNeurons (Additional Table 1). A likened signature of distinct neurodevelopmental disorders may result from shared or interconnected neural gene networks and targetomes of different downregulated brain-specific proteins.
MAGOHB (Magoh Homolog B) (Additional Table 1), is one of the core components of the mRNA splicing-dependent exon junction complex (EJC) which is deposited on nascent spliced transcripts at a fixed position (~24 nt) upstream of splice junctions without specific RNA binding but by means of protein-protein interactions (Bono et al., 2006). MAGOHB,together with its ortholog MAGOH plays a redundant,but essential role in the splicing complex and nonsensemediated mRNA decay, cleavage of growing transcripts in the termination regions, and transport of mature transcripts to cytoplasm (Singh et al., 2013). Several studies have showed that EJC components are essential for brain development and adult brain functioning. Heterozygous mice generated by a ubiquitous conditional deletion ofmagohdevelop microcephaly (McMahon et al., 2014). It is worth noting thatMAGOHBis a top DRG in RSTS neurons (Additional Table 1) and microcephaly is a universal feature of RSTS patients(Hennekam, 2006). Recent studies have confirmed that all EJC protein components, in concert with EJC auxiliary or“peripheral” proteins, impact splicing by suppressing the use of nearby cryptic splice sites (Boehm et al., 2018) and partially spliced transcripts generated by “recursive splicing” (Blazquez et al., 2018). One of the ECJ peripheral proteins, a component of the PASP complex with RNPS1 and SAP18, is the splicing activator Pinin, downregulated in RSTS iNeurons (Additional Table 1). Mechanisms regulating splicing of neuron-specific microexons lead back to ECJ via the combinatorial capacity of ECJ and its peripheral proteins SRSF11 and RNPS1 to interact with the SRRM4 protein, top activator of microexons splicing program (Gonatopoulos-Pournatzis et al., 2018).
PABPC1 (PAPB1) and PAPBC1L are hnRNPs containing four RNA-recognition motifs (Additional Table 2). The first two,RRM1 and RRM2, bind both α-importin and the poly(A) tail of processed mRNA, required for poly(A) shortening which is the first step of mRNA decay and translation initiation. This prevents mRNA from going back into the nucleus. PABPC1 and PAPBC1L are also involved in RNA transport and RNA surveillance pathways (Additional Table 2).
snRNPs
Another set of RSTS iNeurons DRGs includes the Small Nuclear Ribonucleoproteins polypeptides D1, D2, D3, F, G(Additional Table 1), i.e. five of the seven “sm” proteins which make up the structural core shared by the four major RNAprotein assemblies termed uridine-rich U1, U2, U4/U5, U6,small nuclear ribonucleoprotein particles of the eukaryotic pre-mRNA splicing machinery. “Sm” proteins, sharing two evolutionary conserved sequence motifs involved in “sm”protein-protein interactions (Hermann et al., 1995) are critical to the assembly, transport, and integrity of the U snRNPs. It is to be noted that, in particular, members of the spliceosomal U2 snRNPs are, like EJC, proximal partners of the RNPS1 and SRSF11 proteins, co-activators of microexons splicing. For snRNPs too, the RNA-protein interactions are not mediated by RBDs as the spliceosome functional assemblage is driven by the shape and biochemical complementarity between the small nuclear RNAs folded into 3D structures and protein partners (van der Feltz et al., 2012; Hentze et al., 2018). The downregulated snRNP40 and snRNP48 (Additional Table 1) are structural components of U5 and U11/U12 snRNPs,respectively. snRNA-activating protein complex 1 (Additional Table 1) is a transcription factor that, by binding to the proximal sequence element of RNA polymerase II and III promoters, directs both RNA polymerases gene transcription.
Splicing Factors
RSTS neurons DRGs include SRSF3 and SRSF7, two paralogous gene members of the SR-rich family of pre-mRNA splicing factors, which are part of the spliceosome (van der Feltz et al.,2012; Hentze et al., 2018) (Additional Table 1). These RBPs contain an N-terminal RRM for binding RNA and a C-terminal RS domain of consecutive Arginine and Serine dipeptides which facilitates interaction between different splicing factors and other proteins, such as the splicing coactivator Pinin (Additional Table 1), a peripheral EJC protein with an RS domain (Boehm et al., 2018). Besides promoting exon inclusion during AS, they are also involved in mRNA export from the nucleus and in translation.
MBNL3 (Additional Table 1) is a member of the muscleblind family, which mediates pre-mRNA AS regulation as an activator or repressor on specific pre-mRNA targets in antagonism with the splicing activity of the CELF (CUG Elav Family binding protein proteins (Wang et al., 2015). It contains the Zinc knuckle motif (ss-RNA binding zinc finger-CCHC) (Gerstberger et al., 2014b), that can inhibit terminal muscle differentiation(Brinegar and Cooper, 2016) and plays a role in Myotonic Dystrophy pathophysiology.
Downregulated RNA-Binding Proteins Involved in mRNA Transport
Along with several hnRNP family genes, DRGs of RSTS neurons acting in mRNA transport includeNUP35for a nucleoporin which functions as a component of the nuclear pore complex and the transmembraneNDCInucleoporin (Additional Table 3).NUTF2encodes a cytosolic factor that facilitates protein transport into the nucleus and is required for nuclear import of the small Ras-like GTPase which is involved in numerous cellular processes.ENY2(Transcription and Export Complex 2 subunit 2) encodes a multifunctional protein involved in the coupling of transcription with mRNA export. ENY2 associates with a multiprotein complex that remodels chromatin and mediates histone acetylation and deubiquitination and a multiprotein complex implicated in the export of mRNA to the cytoplasm through the nuclear pore, connecting gene expression from transcription to translation (Additional Table 3).
Impaired Ribosome Biogenesis in Rubinstein-Taybi Induced Pluripotent Stem Cell-Neurons
Additional Table 4lists the genes for nucleolar/ribosomal proteins downregulated in RSTS neurons. Ribosomes, the incredibly complex macromolecular machine catalyzing protein synthesis, are composed of four heavily chemically modified non-coding rRNA (5S, 5,8S, 18S, 25/28S) and ~80 structurally distinct proteins. Biogenesis of ribosomes is a process that needs to couple translation of ribosomal proteins to transcription, processing, and modification of rRNAs, and then governs the assemblage of > 200 components (Ojha et al., 2020). Most of the 169 annotated ribosomal proteins lack conventional RBDs and bind directly to rRNA by shape complementarity, operating as RNA-protein complexes,similarly to the vast majority of non-coding RNAs (snRNPs,snoRNPs, telomerase, microRNAs, and lncRNAs) (Hentze et al.,2018).
Prominent for rRNA processing are the small nucleolar RNAs(snoRNAs), a large group of non-coding RNAs which also assist in ribosome assembly, mostly localized in the nucleolus, the site of rRNA biogenesis. Nucleolar proteins and snoRNAs form snoRNPs that mediate post-transcriptional modifications,pseudouridylation, methylation, and cleavage needed for rRNA maturation (Boisvert et al., 2007), contributing to the translational control of gene expression (Sloan et al.,2017). The two key snoRNP complexes are box C/D proteincontaining and box H/ACA protein-containing-RNPs (Watkins and Bohnsack, 2012). Box C/D RNPs catalyze site-specific 2′-O-methylation of rRNAs, snRNAs, and tRNAs in an RNAguided manner: the associated core proteins, together with SNU13 and NOP56 ribonucleoprotein, are the evolutionarily conserved proteins fibrillarin (FBL) and NOP58 (Cervantes et al., 2020), top DRGs in RSTS neurons (Additional Table 4). FBL participates in the first step in processing pre-ribosomal RNA,associated with multiple snRNAs and the U3, U8, and U13 snoRNAs and is the catalytic methyltransferase of the Box C/D snoRNPs mediating the deposition of 2′-O-methylation of~100 residues of rRNA using RNA-RNA base-pairing of one snRNA to direct target sites (Ojha et al., 2020). FBL is equipped with a ribonuclease activity in its glycine/arginine-rich (GAR)domain, conserved in a small group of RNA interacting proteins, such as GAR1 (Additional Table 4). This domain,interacting with phospholipids in the nucleoli, may allow a phase separation (liquid-liquid phase separation) of rRNAassociated proteins containing IDRs, such as fibrillarin and GAR1, thought to be implicated in processing modifications during ribosome production (Guillen-Chable et al., 2020).NOP58 also associates with box C/D U3 and U8 snoRNAs to control specific pre-ribosomal RNA processing steps (Cervantes et al., 2020). Box H/ACA snoRNPs catalyze the isomerization of uridine to pseudouridine using RNA-RNA base pairing to direct target sites: dyskerin, one of the snoRNPs co-factors (together with GAR1, NHP2, and NOP10) is the catalytic subunit (Ojha et al., 2020). TheDKC1-encoded dyskerin pseudouridine synthase1 (Additional Table 4), also plays an active role in telomere maintenance as part of the reverse transcriptase holoenzyme TERT (Additional Table 4), the catalytic core of the ribonucleoprotein complex, having the non-coding TERC RNA as scaffold, that serves as a template for the addition of telomeric repeats to chromosome ends (Roake and Artandi,2020). Other functions of this highly conserved gene are in nucleo-cytoplasmic shuttling, DNA damage response,and cell adhesion.DKC1andTERTmutations are associated with X-linked and autosomal Dyskeratosis congenita (MIM# 305000, MIM # 613989), a severe disorder characterized by bone marrow failure, lung fibrosis and increased susceptibility to cancer. Other rRNA processing RBPs are PseudoUridylate Synthase 7 (PUS7), a component of the chaperone system for assembly and trafficking of snoRNPs,enabling post-transcriptional modification to stabilize RNA secondary molecular structure, and methyltransferase like 1 (METTL1), that mediates the formation of N(7)-methylguanine in tRNAs, mRNAs and microRNAs (Additional Table 4). Biallelic mutations inPUS7are associated with Intellectual Developmental Disorder With Abnormal Behavior,Microcephaly, and Short Stature (IDDABS, MIM # 618342) (de Brouwer et al., 2018).
snoRNA genes are largely found in eukaryotes within the introns of host genes, mainly encoding ribosomal proteins or translation machinery factors, a mechanism allowing coupled synthesis of ribosomal proteins and snoRNAs,which are required for modification of rRNA. Several genes encoding proteins of the small (40 S) or large (60 S) ribosomal subunit are downregulated in RSTS iNeurons (Additional Table 4), includingRPS3andRBL7Awhose mRNAs host one and four snoRNAs, respectively (Ojha et al., 2020). While the loss of a single snoRNA that guides RNA modifications is rarely detrimental, the loss of a snoRNA that directs pre-RNA processing (such as U3 snoRNA) is often lethal (Ojha et al., 2020). The snapshot in RSTS iNeurons of DRGs involved in ribosome biogenesis informs on downregulation of rRNA processing that has been shown to significantly alter ribosome biogenesis and function (Sloan et al., 2017).
Crosstalk between Chromatin Regulation and RNA Processing
The commonalities between the epigenome and the epitranscriptome have been highlighted by the similarity between epigenetic modifications involved in chromatin remodeling and RBPs performance as writers, erasers, and readers of the “epitranscriptomic” marks (Hentze et al., 2018),i.e. the diverse modifications not genetically encoded, but“added on the top” of their RNA partners (Mathlin et al., 2020).
Targeted reviews have discussed the histone marks influencing alternative splicing of neural genes and the underlying mechanisms (Luco et al., 2010, 2011; Hentze et al., 2018;Lipscombe and Lopez Soto, 2019). On the other hand, RBPs have revealed a high level of versatility and some have turned out not only to bind RNA, but also DNA. This is the case with hnRNPA1, hnRNPA2B1, and hnRNPD which are able to interact with single-stranded telomeric DNA (Shishkin et al.,2019) meaning that their decreased amount may lead to reduced telomerase activity (Zhang et al., 2006) or impaired DNA double-strand break repair in the case of hnRNPD which also binds chromatin DNA (Alfano et al., 2019). Technological advances able to capture the chromatin-RNA interactome,such as global RNA interaction with DNA sequencing (GRIDSeq), have detected a consistent number of RBPs RNA binding sites mapping to chromatin-related domains such as the chromodomain and the bromodomain (Hentze et al., 2018; Li and Fu, 2019). The landscape of chromatinassociated RNAs is dominated by pre-mRNAs which are mainly co-transcriptionally spliced and processed by secondary modifications, such as m6A, into mature RNAs. Other chromatin–interacting RNAs include several non-coding RNAs,i.e. snRNAs, snoRNAs, lncRNAs and repeat-derived RNAs, the latter playing a central role in heterochromatin formation. All chromatin-associated RNAs may exert regulatory functions,bridging together transcriptional and post-transcriptional regulation of gene expression by various RNA-mediated feedback and feedforward mechanisms. A key class of RNAs,localized at transcriptional hubs in the 3D genome, are enhancer RNAs (eRNAs) (Li and Fu, 2019), which have been shown to bind the transcriptional coactivator CBP/p300 via its catalytic KAT domain leading to an active H3K27 enhancer state and corresponding changes in gene expression (Bose and Berger, 2017). Acetylation of enhancer-associated regulators features a powerful mechanism to prime networking between chromatin histone modifications and post-transcriptional regulation (Li and Fu, 2019). Interestingly, the snapshot of RSTS iNeurons DRGs sorted to the GO Term Clusters of RNA processing and ribonucleoprotein biogenesis highlights “dual specificity” proteins engaged in the crosstalk between RNA processing and chromatin regulation. Significant examples are the nucleolar FBL, NOP58, and RUVBL1 proteins encoded by top DRGs of RSTS iNeurons (Additional Table 4). The essential FBL methyltransferase responsible for rRNA methylation is also involved in nucleoli-restricted methylation of histone H2A in PolI promoters, where the modified 104 glutamine leads to a weakening of the interaction for the histone chaperone FACT (Facilitator of Chromatin Transcription) complex making PolI transit less impeded by nucleosomes (Tessarz et al.,2014). NOP58 is indicated by proteomics as the prominent interactor of BMAL1, the transcriptional regulator of the circadian rhythm clock, that localizes to the nucleolus where it associates with box C/D snoRNPs (Cervantes et al., 2020).BMAL1-null cells show altered nucleolar morphology and impaired pre-RNA processing, suggesting a non-canonical role of BMAL1 in ribosomal RNA regulation (Cervantes et al.,2020). This “unsuspected” role, shared by PER, the product localized to the nucleolus of another circadian rhythm gene,has been attributed to other classic DNA-binding transcription factors and epigenome regulators that have the ability to directly contact RNA, such as the CCCTC-binding factor CTCF, DNA methyltransferases and other Pol II-associated transcription factors localized in the nucleolus (Li and Fu,2019). RUVBL1 (Additional Table 4), a protein of the ATPases with diverse cellular activities (AAA+) family, has both DNAdependent ATPase and DNA helicase activities and associates with several multisubunit transcriptional complexes and with protein complexes acting in both ATP-dependent remodeling and histone modification. It is part of a chaperone complex aiding the assemblage of C/D box and H/ACA box snoRNPs and their trafficking from the site of transcription through the Cajal bodies to the nucleolus, despite unrelated RNA structures and protein components (Ojha et al., 2020). An additional dual player is SUV39H1 (Additional Table 4), an evolutionarily-conserved protein containing an N-terminal chromodomain, shown to bind RNA (Velazquez Camacho et al., 2017), and a C-terminal SET domain, specific to histone methyltransferases. It trimethylates lysine 9 of histone H3,which results in transcriptional gene silencing (Johnson et al., 2017) and remains associated with heterochromatin by means of an RNA-mechanism mediated by major satellite repeats RNA (Velazquez Camacho et al., 2017). SUV39H1 plays a central role in the establishment of constitutive heterochromatin at pericentric and telomeric regions; loss of function of the gene disrupts heterochromatin formation and may cause chromosome instability.
Beyond these flags, the nexus between CBP/p300 deficiency and a weakened RBPs network in the transcriptome of RSTS iNeurons relies on the finding of reduced expression of the SR-protein SRRM4, the main regulator of the neural-specific microexons AS program, uponCREBBP/EP300depletion(Gonatopoulos-Pournatzis et al., 2018). The top DRGs for the EJC core MAGOHB component and five core proteins of the U snRNPs are upstream partners of the SRRM4 network (Figure 3). Microexons splicing was found to be mis-regulated in the post-mortem brain of one-third of autistic subjects (Irimia et al., 2014) and in patients with autistic and psychiatric disorders (Gandal et al., 2018).CREBBPandEP300are scored in the ASD SFARI database (https://gene.sfari.org/aboutgene-scoring/) in the high confidence category 1 and loss-offunction mutations underlie the neurodevelopmental RSTS syndrome, often characterized by overt ASD or behavior disorder (Ajmone et al., 2018).
Concluding Remarks
We surveyed the landscape of downregulated genes for RBPs in the transcriptome of iNeurons modeling the neurodevelopmental Rubinstein-Taybi syndrome, (Calzari et al., 2020) taking advantage of the fast-growing knowledge of the role of RBPs in the fine tuned post-transcriptional regulation of gene expression. We did not record a single or a few DRGs pointing to a specific gene pathway, but a network of DRGs, encoding defective RBPs connected by their shared roles in alternative splicing and ribosome biogenesis. Dysfunction resulting from the cumulative effect of multiple DRGs is specific to RSTS neurons, even if the cascade of events linking the mutatedCREBBP/EP300genes to the predicted defective RBP-ome profiling is currently unknown. Furthermore, the DRGs of this functionally coherent network encode interacting, often topologically associated, as well as “dual specificity” RBPs linking chromatin to the RBP-ome. This data gives a clue to the disentangling of the increasingly evident disease-associated perturbations of chromatin-RBP-ome cross-talk (Luco et al., 2010, 2011;Gonatopoulos-Pournatzis et al., 2018). The DRGs of RSTS iNeurons comprise genes dysregulated in known neurologic disorders including the recently recognizedHNRNPH1-caused neurodevelopmental syndrome (Pilch et al., 2018;Reichert et al., 2020) which has a striking clinical overlap with Rubinstein-Taybi, possibly due to the numerous reciprocal interactions that deregulated RBPs engage forming RNPs with partially shared targetomes. The cross disorders marks, such as the presence, independent of mutations, of different RBPs sequestered in RNA foci in patients with neurodegenerative diseases, raise the challenge of discriminating their specific etiological contribution and enhance the development of therapeutic strategies with the potential to disrupt protein-RNA interfaces altered in disease states (Harrison and Shorter, 2017). As every RBP can control alternative splicing choices and may participate in interconnected pathways of ribonucleoprotein complex biogenesis, a high number of missplicing events is likely driven by the large set of downregulated RBPs in CBP/p300 defective iNeurons. Our transcriptome analysis focused on the differential expression of transcripts from annotated coding genes, while isoform level changes that capture the largest effects of diseased neurons (Gandal et al., 2018) were not addressed. A further layer of deleterious consequences, i.e. altered isoform expression, is predicted in RSTS iNeurons based on studies highlighting that CBP/p300 promotes the expression of the SRRM4 regulator, top hit of the highly conserved neuronal microexon splicing, deranged in Autism and psychiatric disorders (Gandal et al., 2018). Reduced CBP/p300 activity is not only at the roots of the rare Rubinstein-Taybi syndrome,but also implicated in several neurodegenerative conditions suggesting strategies for reinstatement of KAT activity based on epidrugs, such as histone deacetylase inhibitors (Valor et al., 2013). iPSC-derived cortical neurons from patients with diverse neurodegenerative diseases continue to provide insights into pathomechanisms underlying neurodegeneration and a platform for drug screening (Lines et al., 2020). The use of class I histone deacetylase inhibitors applied to forms of frontotemporal dementia caused by mutation of theGRNgene has been proposed as a promising therapeutic strategy to counteract the causative progranuline haploinsufficiency(She et al., 2017). Lastly, CBP and p300 are very large proteins that contain seven folded domains, but the regions outside these globular domains, accounting for about 60% of the sequence, are predicted to be intrinsically disordered and a disordered, autoinhibitory loop of ~60 residues is embedded in the KAT domain too (Dyson and Wright, 2016). The hub position of CBP/p300 in chromatin regulation is well attested by its huge interactome/acetylome (Bedford et al., 2010;Weinert et al., 2018). It remains to be seen whether CBP/p300 structural features, which resemble RBPs common features and pathomechanisms driving diseases with disrupted cognition, might account for a hub position of CBP/p300 in the RBP-ome.
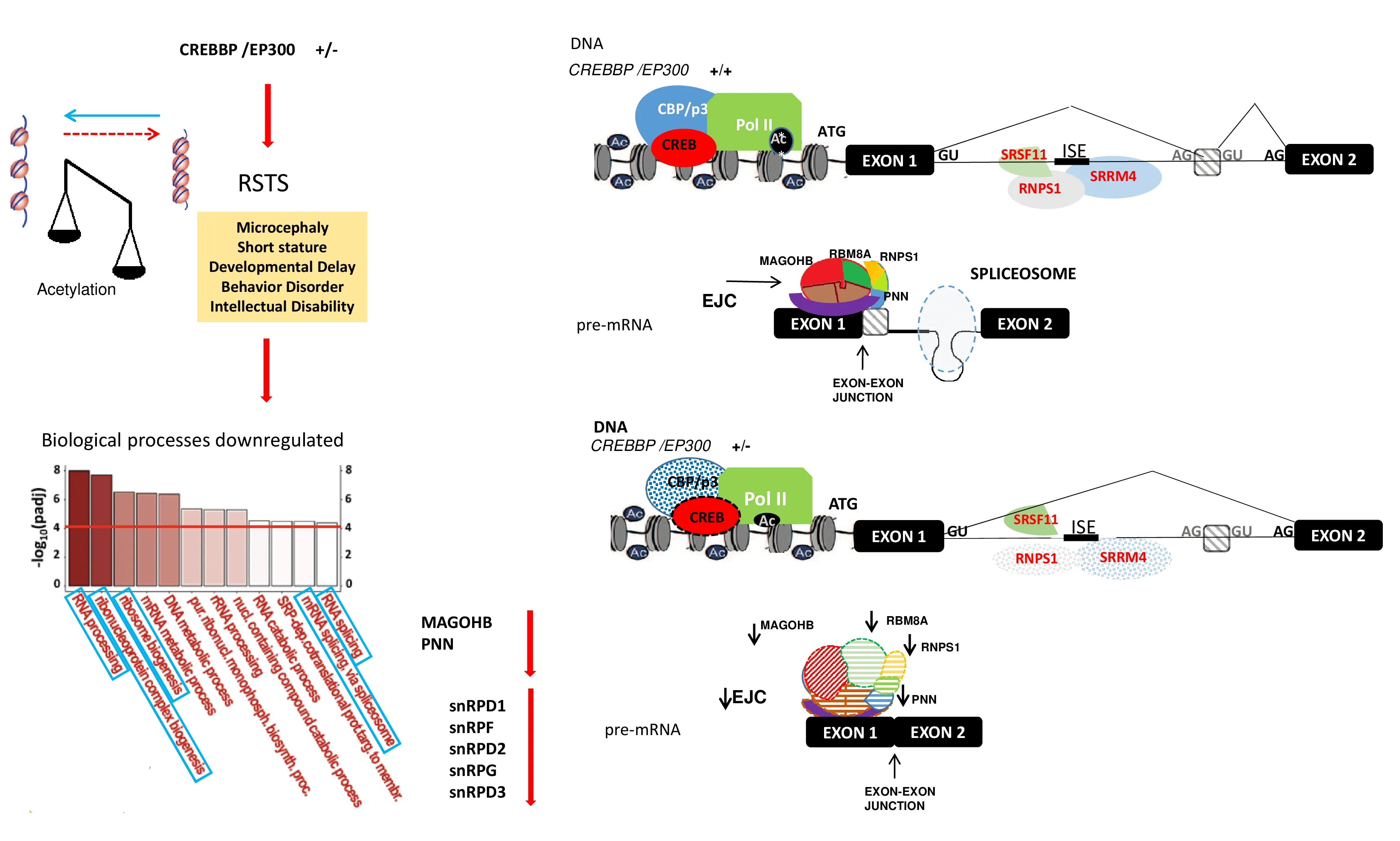
Figure 3|Impact of the acetylation defect in Rubinstein-Taybi syndrome (RSTS) on neuralspecific microexons splicing.
Author contributions:All authors contributed to all-round analyses of the genes downregulated in RSTS neurons by literature datamining and access to(epi)genomic, cell biology and transcriptomic databases. Cross comparison of the same/ortholog genes in human and mouse neuronal systems reported in the literature was addressed by extensive discussion on bioinformatics (LC),cell biology (VA) and human and medical genetics (SR and LL) aspects. LL drafted the manuscript that was revised upon general discussion. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was supported by Italian Ministery of Health RC 08C921 to LL, Istituto Auxologico Italiano, IRCCs.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional Table 1: RNA-binding proteins genes mainly involved in alternative splicing downregulated in Rubinstein-Taybi syndrome induced pluripotent stem cell neurons.
Additional Table 2: hnRNPs downregulated in Rubinstein-Taybi syndrome neurons, acting in the regulation of alternative splicing: structural motifs,functions and link to disease.
Additional Table 3: Genes for RNA binding proteins downregulated in Rubinstein-Taybi syndrome induced pluripotent stem cell neurons involved in mRNA transport and translation initiation.
Additional Table 4: Genes for pre-rRNA processing and ribosome biogenesis downregulated in Rubinstein-Taybi syndrome induced pluripotent stem cell neurons.

- 中国神经再生研究(英文版)的其它文章
- Research advances on how metformin improves memory impairment in “chemobrain”
- Dendritic spine density changes and homeostatic synaptic scaling: a meta-analysis of animal studies
- Optogenetic activation of intracellular signaling based on light-inducible protein-protein homo-interactions
- Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease
- Growth differentiation factor 5: a neurotrophic factor with neuroprotective potential in Parkinson’s disease
- The promise of neuroprotection by dietary restriction in glaucoma