
Small scale adeno-associated virusvector production for preclinical gene delivery based on chloroform precipitation
2022-06-13 08:29MarcusDavidssonAndreasHeuer
中国神经再生研究(英文版) 2022年1期
Marcus Davidsson, Andreas Heuer
Gene therapy aims to introduce genetic information into a cell-type of interest to replace, correct, silence, or modify defective genes. Gene therapy in its broadest sense can theoretically prevent, halt, or cure any condition that affects mankind. In addition to that, the introduction and/or manipulation of genes is one of the major research areas in biological sciences, aimed to deepen our knowledge on how biological systems work.Scientific advances have made it possible to induce changes ranging from manipulations of large stretches of the genome to the change of single nucleotides. The gold-standard vehicles to bring this genetic information into the target cells are viral vectors, amongst which the adeno-associated virus (AAV) is the most commonly used. AAV-vectors are small single stranded DNA viruses that naturally infect cells in humans and other primate species, thereby making them a perfect candidate for genetherapy. In contrast to retroviruses, such as lentiviral vectors, AAVs are replication deficient and do not integrate into the host genome thus reducing the risk of insertional mutagenesis.Furthermore, AAVs are currently not known to cause any diseases in humans. The AAVs have an excellent biosafety profile and approximately 80–90% of the human population is already carrying the virus. There are currently about 13 naturally occurring variants known (serotypes),each with a different tropism profile to specific cell/tissue types (Vance et al., 2015). In the clinic, AAV-vectors have been shown to achieve stable and long-term transgene expression.Currently over 215 clinical trials are either ongoing or completed using the AAV-vector platform (www.clinicaltrials.gov), targeted to a wide range of disorders such as haemophelia A and B, Duchenne muscular dystrophy, spinal muscular atrophy, Leber congenital amaurosis,but also neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease(Ginn et al., 2018). Two AAV-based drugs have already achieved FDA approval, Luxturna in 2017 for rare inherited retinal dystrophy and Zolgensma in 2019 for spinal muscular atrophy.For preclinical research, the AAV-vector is the workhorse to achieve transgene expression in different tissue types in a multitude of biological systems. By use of cell-type specific enhancers/promoters (Blankvoort et al., 2020),tissue selective capsid variants (Davidsson et al., 2019; Weinmann et al., 2020), or conditional expression systems (e.g., Crerecombinase or tetracycline) (Chtarto et al.,2003), these vectors have been used for tissue labelling and circuit tracing (e.g., INTRSECT)(Fenno et al., 2014), cell activity manipulation(e.g., Optogenetics, Chemogenetics) (Aldrin-Kirk et al., 2018), activity monitoring (e.g.,calcium indicators) (Chen et al., 2013), disease modelling (e.g., protein overexpression) (Kirik et al., 2002), and gene editing (e.g., CRISPR/Cas) (Jinek et al., 2012). Future work on AAV development is largely focussed on achieving higher selectively in cell type specificity(tropism), transduction efficiency (transgene expression), avoiding host immune responses,and increasing the packaging capacity of the viral capsid. Additionally, increasing the production yield is a challenge for the future as the demand has increased in recent years as very large volumes of high genome copy (gc)-titer vector are used in clinical applications and preclinical systemic injection studies. To achieve cell specific tropism, increased and cell specific transgene expression, larger cargo capacity, as well as increased production yield, several lines of research are ongoing focussing on i) capsid development, ii) expression cassette, and iii)production methods (Figure 1, top). New natural and synthetic AAV-capsid variants are constantly being identified and developed.Development of new capsids is based on rational design, directed evolution (e.g. CREATE)(Deverman et al., 2016), barcoding (e.g. BRAVE)(Davidsson et al., 2019), or computer guided generation for new peptide/capsid configurations (Ogden et al., 2019). These strategies have resulted in many remarkable improvements such as the generation of retrograde AAVs (AAV2-retro; MNM004/008)(Tervo et al., 2016; Davidsson et al., 2019) and AAVs that can cross the Blood Brain Barrier(PHP.B) (Deverman et al., 2016). Larger cargo capacity can be achieved by adding additional viral capsid protein units or by splitting the delivery over several vectors. Stronger and faster transgene expression as well as cell type specificity can be achieved by novel combinations of promotor-enhancer elements(Blankvoort et al., 2020), codon optimization of the transgene, transcription-termination sequences (e.g., polyA) as well as the introduction of protein stabilizing sequences(e.g., WPRE). Another approach for faster and increased expression was the development of a double-stranded AAV vector system. Although it reduces the cargo capacity by approximately half, the transgene expression does not have to go through the second strand synthesis, which is a rate-limiting process in the transgene expression. Improvements in production methods are mainly focusing on vector yield to accommodate for the increased demand in high gc-titer AAVs for preclinical and clinical approaches. Large volumes are needed for several approaches, ranging from iv injections for circuit tracing (PHP.B), preclinical primates as well as clinical trials, where titers in the range of 1 × 1014gc/mL per kg of body weight are becoming the norm (Li and Samulski, 2020).AAV production methods rely on a transfectionbased method by which the two or three necessary plasmids are provided separately to a producer cell line. There are currently four different production systems: HEK 293T transfection, stable cell lines, the HSV system,and the baculovirus system using Sf9 cells. The main challenge is the scalability of the given approach, as most methods were developed to produce small to medium quantities. The HEK293T system is the most commonly used approach and relies on the delivery of pAAVrepcap, pAd-helper, and the pAAV-transgene(containing the ITRs) in three separate plasmids. A two plasmid system where both helper plasmids are merged into one plasmid has been developed as well (Grimm et al.,2003), which increases the likelihood of cotransfection. Traditionally, T175 flasks or comparable cell culture dishes are used for production and scaled up approaches use roller-bottles or fixed bed reactor systems (in combination with stable cell lines). Production methods using suspension culture could be scaled to using large volume bioreactors(Grieger et al., 2016) for clinical trials. There are several interesting developments such as the generation of producer cell lines, packaging cell lines, and novel cell lines for free-floating cell culture. Although the HSV system can be used to produce AAV vectors, the low risk of replication competent HSV through recombination cannot be completely excluded,therefore an additional step of inactivation/removal/validation must be taken. The baculovirus/insect cell system is based on Sf9 cells which are grown in suspension cultures where different baculoviruses deliver the essential genes for AAV production. Currently,production of AAV-vectors is based on either a form of density gradient using iodixanol or caesium chloride, different versions of chromatography purifications, or on our currently presented chloroform precipitation method (Davidsson et al., 2020; Negrini et al.,2020). Although large volume batches of AAV vectors are needed for preclinical and clinical trials, current production methods are not very economical or time saving, when testing several different variants. When testing e.g. different promoters, only small volumes are needed to compare which promoter achieves the desired expression levels in the target tissue. The iodixanol-gradient purification results in volumes of about 200 µL AAV vector, takes 9 days to complete, and requires specialized equipment such as ultracentrifuges and pumps,which are costly infrastructure that might not be available. Our novel PEG precipitation and chloroform extraction method relies on standard cell culture equipment and negates the use of ultracentrifugation. The methods differ mainly in the time of purification, yield,and purity of final AAVs. In our recently publish protocol, we show a fast and efficient in-house production method that can be carried out in any normally equipped lab (with the appropriate safety classification). We have shown that this method, compared to standard Iodixanol purification, produced vectors with comparable purity and packaging efficacy (>90% packaged particles). We compared AAVvectors of both production methodsin vitroon their ability to infect cells in culture andin vivoon their ability to infect cells after injection into the rat midbrain. The vectors of both productions were equipotent in their ability to transduce cellsin vivoandin vitroand neither method elicited an inflammatory response that went beyond the local inflammation due to the surgical procedure (Davidsson et al., 2020). The choice of production method should be focused on the final goal of the product produced. Our novel protocol provides a cost-benefit ratio which is favorable for preclinical studies to test a multitude of factors. When considering the costs of consumables and the experimentertime per AAV vector batch produced, especially when the residual AAV vectors from comparisons are not used further. As the chloroform protocol allows for fast production of multiple batches of small volumes (25–40µL), it is an ideal method for in-house testing,before committing to any of the production methods that yields larger volumes of purified AAVs (Figure 1, bottom). The fast, small scale method is superior when it comes to multiple parameter testing, such as different capsids,promoters, transgenes, termination signals (i.e.,polyAs) and stabilizing elements (i.e., WPREs).This provides a significant advantage for the preclinical evaluation, as many variants can be quickly compared side-by-side. We have used the chloroform method for both naturally occurring serotypes and engineered capsids,generating hundreds of batches. Furthermore,by using the chloroform protocol, it is possible to harvest and purify > 20 virus batches in a single day, which would take considerably longer using a standard iodixanol protocol.Despite the great promise that gene-therapy holds, the main limitation to achieve rapid progress in the field is the time and financial resources it takes to test various constructs.Although we anticipate that recently developed techniques such as barcoding or computerguided generation of new variants will significantly speed up development and narrow down targets,in vivotesting of selected constructs will remain the gold standard for the foreseeable future. With our small-scale protocol we are not only able to generate multiple constructs in a shorter timeframe, but also open the field for many research groups that do not have access to specialized equipment. Gene therapy based on AAV vectors and non-viral nanoparticles will continue to provide unique opportunities to correct faulty genes and improve human health at a rapid pace. Our excitement by this approach is not only justified by the current technological state of the art, but also by the promises that the future holds. Precise control over genes such as base and prime editing has the potential to revolutionize personalized medicine. Our method helps with that process, and could speed up experimental therapeutic options,that hopefully can transition into successful clinical therapeutic modalities.
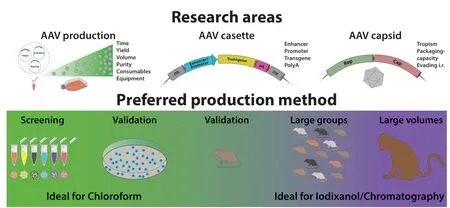
Figure 1|AAV research and production.
MD and AH would like to thank David Marmion from Barrow Neurological Institute (Phoenix,Arizona, USA) for his valuable input on this manuscript.
The present work was supported by the Royal Physiographic Society of Lund(Kungliga Fysiografiska sällskapet), the royal Swedish academy of sciences (Kungliga Vetenskapsakademien) and the Per-Eric och Ulla Schybergs foundation (to MD), the Swedish Research Council (Vetenskapsrådet),the Parkinsons foundation (Parkinsonsfonden),the Åhlens foundation (Åhlensstiftelsen), the Jeanssons foundation (Jeanssons stiftelser) as well as the Crafoord foundation (Crafoordska stiftelsen) (to AH).
Marcus Davidsson*, Andreas Heuer*
Molecular Neuromodulation, Department of Experimental Medical Sciences, Lund University,Lund, Sweden (Davidsson M)
Behavioural Neuroscience Laboratory, Department of Experimental Medical Sciences, Lund University,Lund, Sweden (Heuer A)
*Correspondence to:Marcus Davidsson, PhD,marcus.davidsson@med.lu.se; Andreas Heuer,PhD, andreas.heuer@med.lu.se.
https://orcid.org/0000-0003-0233-0516(Marcus Davidsson)
https://orcid.org/0000-0003-0300-7606
(Andreas Heuer)
Date of submission:December 22, 2020
Date of decision:January 19, 2021
Date of acceptance:February 20, 2021
Date of web publication:June 7, 2021
https://doi.org/10.4103/1673-5374.314309
How to cite this article:Davidsson M, Heuer A(2022) Small scale adeno-associated virus-vector production for preclinical gene delivery based on chloroform precipitation. Neural Regen Res 17(1):99-100.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under theterms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Janne Koskimäki, University of Turku, Finland.

- 中国神经再生研究(英文版)的其它文章
- Genes for RNA-binding proteins involved in neuralspecific functions and diseases are downregulated in Rubinstein-Taybi iNeurons
- Research advances on how metformin improves memory impairment in “chemobrain”
- Dendritic spine density changes and homeostatic synaptic scaling: a meta-analysis of animal studies
- Optogenetic activation of intracellular signaling based on light-inducible protein-protein homo-interactions
- Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease
- Growth differentiation factor 5: a neurotrophic factor with neuroprotective potential in Parkinson’s disease