
Fibrosis as a common trait in amyotrophic lateral sclerosis tissues
2022-06-13 08:29SavinaApolloniNadiaAmbrosi
中国神经再生研究(英文版) 2022年1期
Savina Apolloni, Nadia D’Ambrosi
Amyotrophic lateral sclerosis (ALS) is a highly aggressive adult-onset neurodegenerative disease caused by the progressive loss of upper and lower motor neurons. Clinically,it causes irreversible muscle atrophy and spasticity, leading to death due to respiratory failure, usually within 2–5 years after the first symptom onset. Approximately 85% of ALS cases are classified as sporadic, while the remaining 15% are of familial origin, but the overall clinical and molecular features of the disease are almost undistinguishable in the two forms. The majority of familial ALS cases are caused by pathogenic variants ofC9orf72,SOD1,TARDBP,FUS,ANGandOPTNgenes that are inherited by a Mendelian pattern and display high penetrance (Kiernan et al., 2020).
Although significant progress has been made in the last decade in revealing new genes and mechanisms contributing to the disease, ALS still lacks effective therapies.It is now clear that the pathology raises not only from gene loss-of-function, rather from gain-of-toxic functions, that multiple altered cellular pathways contribute to the pathological outcomes and that the disease is non-cell autonomous. A number of pathogenic mechanisms that are likely to be interconnected have been detected in different cell types in addition to motor neurons, and include the formation of protein aggregates, RNA dysmetabolism,cytoskeletal rearrangements, increased oxidative stress, mitochondrial dysfunction and metabolic imbalance (van Es et al.,2017). Likewise, inflammation is a shared pathophysiological response occurring in different regions: in the central nervous system (CNS) neuroinflammation by microglia and astrocytes precedes and aggravates motor neuron death; peripheral immune cells are found in the CNS and muscle,contributing to motor neuron demise and to muscle atrophy; skin fibroblasts exhibit proinflammatory phenotypes.
We propose here that fibrosis, an event whose initiation and progression are intimately dependent on inflammation,may be considered a pathogenic molecular mechanism arising in different organs in ALS. In light of this, we suggest that the systemic control of fibrotic reactions can ameliorate the disease outcome.Fibrosis is a process of uncontrolled tissue reorganization that begins as a response to limit and repair damage, i.e., wound healing, but evolves over time leading to distortion of tissue architecture and creating a hostile environment to cell regeneration.Literature on the non-cell autonomous aspects of ALS in the spinal cord and cerebral cortex suggests the presence of interspersed fibrotic scars closely bordered by glial scars. The fibrotic components are mainly constituted by endothelial cells,inflammatory immune cells, myofibroblasts and extracellular matrix (ECM) molecules such as proteoglycans, fibronectin, collagen IV and connective tissue growth factor(CTGF/CCN2) and are accompanied by dysregulation of matrix metalloproteases(MMPs) and thickening of the perineuronal networks. Accordingly, several markers that can suggest the establishment of a fibrotic environment, as transforming growth factor-β1 (TGF-β1), signal transducer and activator of transcription 3 (STAT3), vimentin,and α-smooth muscle actin (α-SMA),increase in affected tissues of the CNS of ALS patients and inin vivomodels of the disease(D’Ambrosi and Apolloni, 2020). Moreover,we have found that S100A4 (also called fibroblast-specific protein 1), a protein deeply involved in cell migration and epithelial-tomesenchymal transitions, as well as a key player in fibrotic diseases, is upregulated in the spinal cord of ALS models at an early symptomatic phase of the disease. We have also demonstrated that S100A4 inhibitionin vitroby its transcriptional inhibitor niclosamide, a well-known anti-fibrotic agent, leads to decreased production of several pro-inflammatory markers, including the mammalian target of rapamycin and nuclear factor-κB, and to reduced cell migration (Serrano et al., 2019). Remarkably,high S100A4 levels in patients’ serum strictly correlate with disease progression in several fibrotic diseases and are considered a useful biomarker for diagnosis and monitoring disease outcome (Li et al., 2020).
In ALS, besides the nervous system also peripheral tissues show marked signs of fibrosis and inflammation that can be associated with tissue degeneration. For instance, alterations in skeletal muscles,as a consequence of early pathological denervation, contribute to ALS progression,and muscle fibrosis is a pathological hallmark of the disease. Indeed, skeletal muscles from patients with ALS show elevated levels of collagen type XIX alpha 1 protein that have been positively correlated with increased mortality and rapid progression of the disease (Calvo et al., 2018). Moreover,the skeletal muscle of symptomatic SOD1-G93A mice displays an upregulation of the pro-fibrotic CTGF/CCN2, accompanied by an excessive deposition of ECM molecules.Importantly, the inhibition of CTGF/CCN2 with the monoclonal neutralizing antibody FG-3019 in these mice can reduce fibrosis and muscle atrophy, also improving motor performance and neuromuscular junction integrity (Gonzalez and Brandan, 2019).The same group has also reported that other ECM components and fibrotic genes,as platelet-derived growth factor (PDGF)receptor α, transcription factor 4, and α-SMA, are augmented in skeletal muscles of symptomatic SOD1-G93A mice, as well as the levels of pro-fibrotic factors TGF-β1 and Smad3, all converging to fuel a fibrotic response in this tissue (Gonzalez et al.,2017). Finally, in both SOD1-G93A mouse model and in skeletal muscle from ALS patients, denervation elicits an aberrant activation of STAT3-interleukin-6 signaling in the fibro-adipogenic progenitors, the main producers of ECM constituents, leading to muscle fibrosis and atrophy. Consistently, the inactivation of STAT3 has been demonstrated as a successful therapeutic strategy capable of improving muscle degeneration and fibrosis in ALS mice (Madaro et al., 2018).Altogether, these data strongly indicate that targeting fibrotic pathological processes could represent a valuable approach to counteract skeletal muscle waste occurring during ALS.
A common cause of death in ALS, in addition to respiratory paralysis, is sudden heart failure, indicating a possible cardiomyopathy involvement in the disease. Through the employment of cardiac magnetic resonance imaging, it has been demonstrated that ALS patients display structural myocardial defects and that a trend toward late gadolinium enhancement, accounting for organ fibrosis, occurs in a percentage of patients. In relation to this aspect, cardiac fibrosis may be considered a risk factor for heart failure in patients with ALS (Rosenbohm et al., 2017). Moreover, ALS patients also show hepatic abnormalities and atrophy,with granular inclusions and lymphocytic infiltration. Consistently, in the liver of SOD1-G93A mice, the expression of proteins related to inflammation, oxidative stress and fibrosis increases with disease progression.Indeed, histone deacetylase 4, DNA-damageinducible 45α and PDGFβ are augmented in the early phases of the disease, indicating that liver dysfunction, mediated by increased inflammation and oxidative stress, as well as by fibrosis-related processes, precedes muscle atrophy symptoms (Lee and Yang,2018).
Finally, a tissue showing important signs of fibrosis in ALS is represented by the skin.Indeed, many dermis changes have been reported in ALS patients. Several collagen abnormalities, related to collagen levels,density and fiber diameter have been accounted. According to what happens in the CNS, also the levels of MMP-9 are increased in the skin, possibly in relation to the abnormal collagen deposition, and for these reasons the level of MMP-9 in the skin of patients suffering of ALS has been proposed as a potential biomarker.Furthermore, hyaluronic acid, a key component of the ECM pertaining to the family of glycosaminoglycans, and which is implicated in wound healing processes,inflammation and scarring is increased in the skin of ALS patients. Finally, the overexpression of the glycoprotein laminin 1 at the basement membrane has also been detected in patients. Similarly to what occurs in the CNS, the skin displays a high level of cytokines, such as interleukin-6 and tumor necrosis factor-α. Both these molecules are pro-inflammatory factors that may play roles in wound healing by regulating cell proliferation, differentiation and migration (Paré and Gros-Louis, 2017).Altogether, these results suggest that ECM alterations are common marks in the skin of ALS patients, supporting the idea of a skin involvement in the disease. It has been proposed that these pathological hallmarks may mirror disturbances in the CNS and peripheral nerves. Dermic fibroblasts in particular are widely used as a model system to study different altered cellular pathways that occur in the CNS. Like cells of neural origin, they show alterations in metabolism,protein homeostasis, DNA-damage response,autophagy and in pro-inflammatory phenotypes that can be related to fibrotic pathways (Parè and Gros-Louis, 2017;Riancho et al., 2020). In particular, we have shown that fibroblasts deriving from patients carrying SOD1 pathogenic variants express high levels of S100A4, mammalian target of rapamycin and nuclear factor-κB, indicating an alteration in the inflammatory and fibrotic events (Serrano et al., 2019).
The results discussed here open a new and exciting field in the study of fibrosis as a common trait in different ALS tissues(Figure 1). Fibrosis occurs after a necessary response to functional impairments, but the development of such a massive response characterized by the substitution of parenchymal tissue with ECM might turn to be a negative event, particularly because it sustains chronic inflammation and it impedes any attempt of regeneration.
On these grounds, the possible treatment with anti-fibrotic compounds that may attenuate inflammation, secretion of molecules such as TGF-β1 and tumor necrosis factor-α, activation of pathways involving STAT3 and the excessive deposition of ECM components, offers a chance to improve the disease outcome acting on multiple organ dysfunctions, and should be considered in a multi-targeted therapeutic approach to counteract ALS.
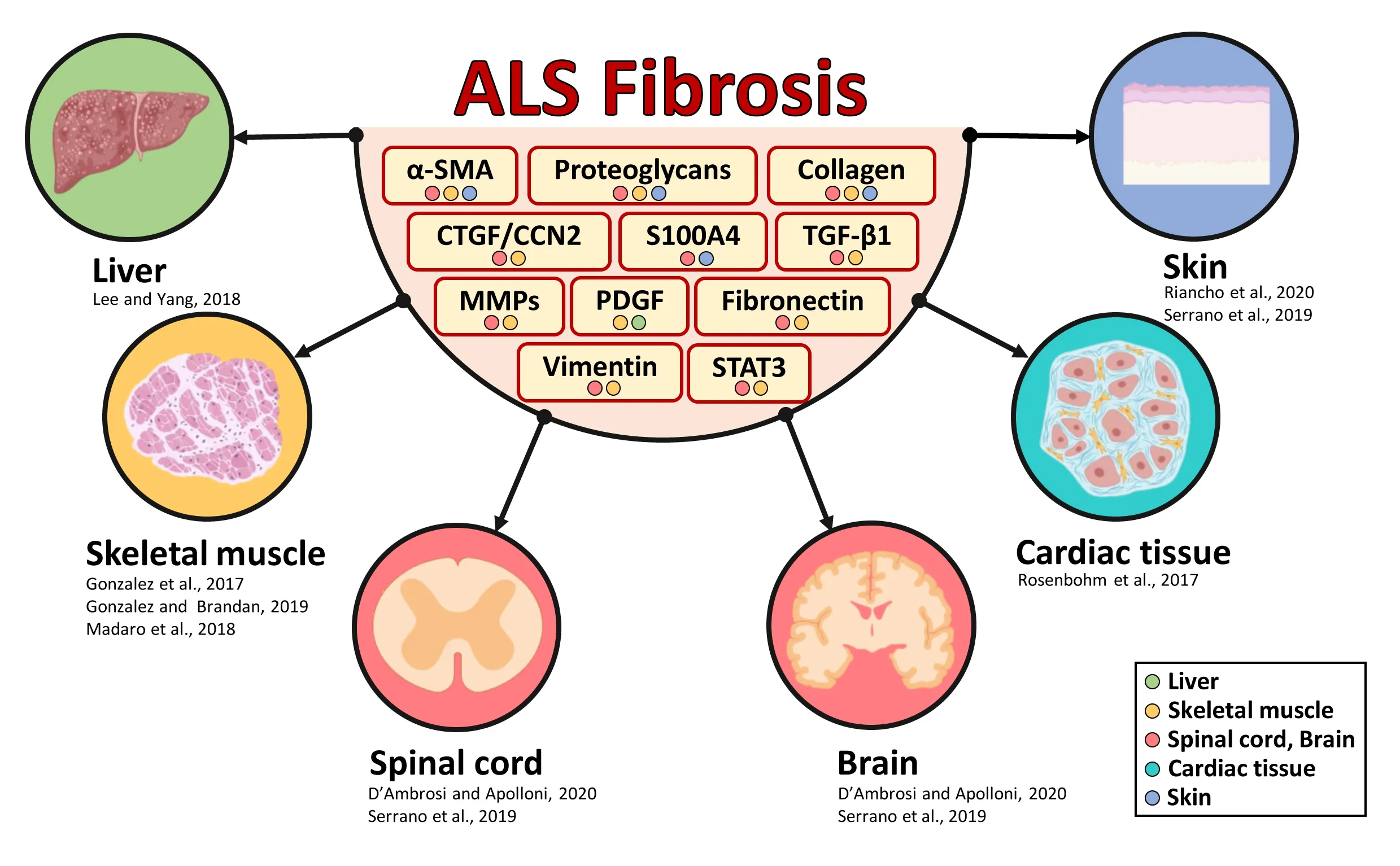
Figure 1|Schematic representation of fibrosis involvement in amyotrophic lateral sclerosis (ALS).
Savina Apolloni*, Nadia D’Ambrosi*
Department of Biology, University of Rome Tor Vergata, Rome, Italy
*Correspondence to:Savina Apolloni, PhD,
savina.apolloni@uniroma2.it; Nadia D’Ambrosi,PhD, nadia.dambrosi@uniroma2.it.
https://orcid.org/0000-0002-5782-1665(Savina Apolloni);
https://orcid.org/0000-0002-6646-7653(Nadia D’Ambrosi)
Date of submission:November 10, 2020
Date of decision:December 26, 2020
Date of acceptance:February 22, 2021
Date of web publication:June 7, 2021
https://doi.org/10.4103/1673-5374.314302
How to cite this article:Apolloni S, D’Ambrosi N(2022) Fibrosis as a common trait in amyotrophic lateral sclerosis tissues. Neural Regen Res 17(1):97-98.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:David Sassoon, Inserm and UCSF/VA Hospital, France.

- 中国神经再生研究(英文版)的其它文章
- Genes for RNA-binding proteins involved in neuralspecific functions and diseases are downregulated in Rubinstein-Taybi iNeurons
- Research advances on how metformin improves memory impairment in “chemobrain”
- Dendritic spine density changes and homeostatic synaptic scaling: a meta-analysis of animal studies
- Optogenetic activation of intracellular signaling based on light-inducible protein-protein homo-interactions
- Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease
- Growth differentiation factor 5: a neurotrophic factor with neuroprotective potential in Parkinson’s disease