
Ferroptosis: copper-iron connection in cuprizone-induced demyelination
2022-06-13 08:29PriyaJhelumSamuelDavid
中国神经再生研究(英文版) 2022年1期
Priya Jhelum, Samuel David
Redox active metals such as iron, copper,zinc, and manganese play important roles in promoting a variety of biochemical reactions essential for cellular function. This is made possible by the ability of these metals to accept and donate electrons. Iron in the form of iron-sulfur clusters and heme plays a key role in adenosine triphosphate generation in mitochondria as well as numerous other enzymatic reactions. On the other hand,disruption in the normal homeostatic levels of these metals, either excess or reduction, results in damage to cells and to tissue pathology.Excess copper accumulation in the central nervous system (CNS) in Wilson’s disease results in damage to the basal ganglia, and excess iron deposition in the CNS in aceruloplasminemia results in damage to various regions of the brain and retina. Such damage is thought to be induced by free radicals generated via Fenton chemistry. Here, we focus on our recent work that revealed a role for ferroptosis, a form of iron-mediated cell death, in cuprizone-induced oligodendrocyte loss and demyelination(Jhelum et al., 2020). Cuprizone is a copper chelator that induces demyelination in experimental animals and is widely used to study demyelination and remyelination in the CNS (Zhan et al., 2020), often in the context of multiple sclerosis (MS), the prototypical demyelination disease in humans. Another key finding in this work is the potential role of ferritinophagy in ferroptosis, i.e., the release of iron from cytosolic ferritin (Dixon et al., 2012;Mancias et al., 2014). This work also shows the link between copper and iron metabolism, in which chelating copper leads to dysregulation of iron metabolism. Importantly, ferroptosis may also play a role in other neurological conditions and deserves further study.
Iron deposition occurs in the CNS in aging and a wide range of neurological disorders that include MS, amyotrophic lateral sclerosis,Parkinson’s disease, stroke, and spinal cord injury. In hemorrhagic stroke and CNS trauma,iron deposition is immediate as much of the iron is derived from hemoglobin in red blood cells (RBC). Each human RBC contains about 270 million molecules of hemoglobin each containing four heme groups with an iron at its center. Thus, each RBC is loaded with over a billion atoms of iron. Heme itself is also highly cytotoxic. The large number of RBCs at the site of hemorrhage leads to tremendous accumulation of iron in macrophages in the affected tissue (Rathore et al., 2008). Excess iron gets stored in cytosolic ferritin. In other neurological conditions in which bleeding does not occur, iron accumulation occurs over a longer period due to dysregulation of cellular iron metabolism and iron released from dying cells. Nevertheless, iron accumulation in these conditions (e.g., MS, amyotrophic lateral sclerosis) can be quite substantial and can be detected by histochemical methods. Excess iron in cells in pathological conditions, can be mobilized from ferritin in which it is generally stored and generate free radicals via the Fenton reaction. It would also appear that small,transient changes in the level of intracellular bioactive iron may also induce iron-mediated cytotoxicity.
Ferroptosis was discovered in 2012, first in the context of work on cancer cellsin vitro(Dixon et al., 2012) and subsequently shown to be implicated in other conditions including some neurological conditions, such as Parkinson’s disease, stroke and CNS injuries (Guiney et al., 2017; Stockwell et al., 2017; Magtanong and Dixon, 2018; Zhang et al., 2019).Ferroptosis is an iron-mediated, non-apoptotic form of cell death. It differs biochemically and morphologically from apoptosis and other forms of cell death (necroptosis and pyroptosis). Several features of ferroptosis are worth noting (Dixon et al., 2012; Dixon and Stockwell, 2014). One is its dependency on iron as seen by the ability of iron chelators such as deferoxamine to block ferroptosis. Therefore,iron accumulation in tissues or dysregulation of cellular iron metabolism leads to increases in bioactive iron that can trigger ferroptosis.Another important feature of ferroptosis is the generation of lipid radicals. Associated with this, is the inability of endogenous mechanisms to scavenge lipid radicals. Reduction or depletion of glutathione (GSH), an antioxidant,results in iron-dependent accumulation of lipid radicals and loss of cell viability. Mechanisms that reduce GSH levels will therefore induce ferroptosis. These include impairment or reduction of expression of system Xc (or xCT),a cystine antiporter, which would lead to reduction in cystine, needed for synthesis of GSH. Another feature of ferroptosis is reduction of glutathione peroxidase 4 (GPX4), an enzyme that helps GSH neutralize lipid radicals. It is noteworthy that small molecules such as ferrostatin-1 and liprostatin-1, which scavenge lipid radicals can prevent ferroptosis-mediated cell death. Therefore, knowing whether ferroptosis contributes to the pathology of a neurological disorder will determine if such conditions are suitable for drug therapies that specifically target ferroptosis.
A PubMed search revealed that 930 papers on cuprizone were published in the past 55 years since it was first shown to cause damage to the CNS. Interestingly, there were few papers until the turn of this century, with only 4 papers published in the year 2000 but the numbers rose sharply to 119 papers in 2019. There is therefore an increasing interest in the use of the cuprizone model. Although 180 papers dealt with mechanisms underlying demyelination and remyelination, the precise mechanism that trigger oligodendrocyte cell death and demyelination has remained elusive. We became interested in whether copper chelation by cuprizone will dysregulate iron homeostasis because of the long-standing work in our lab showing that ferroxidases (ceruloplasmin and hephaestin) play an important role in iron eラux or export from cells, and that lack or deficiency in these ferroxidases leads to iron accumulation in the CNS (Jeong and David, 2006; Schulz et al.,2011). These two ferroxidases are homologues that differ mainly in their membrane-anchoring regions. They are among the most copper-rich enzymes, each molecule containing 6 coppers,which are essential for electron transfer reactions and ferroxidase activity (Bento et al.,2007). In the CNS, ceruloplasmin is expressed in astrocytes, while hephaestin is expressed in oligodendrocytes. Ceruloplasmin knockout mice first showed presence of iron accumulation in astrocytes at 18 months of age (Jeong and David, 2006), while hephaestin mutant mice show iron deposition in oligodendrocytes by 2 months of age but it may likely occur even earlier (Schulz et al., 2011). These findings show that there are differences in iron cycling and utilization in these two glial cell types.Iron deposition in CNS cells can be detected in these conditions by iron histochemistry, as well as by increased expression of ferritin, an iron storage protein, which is a sensitive indicator of iron. It was therefore reasonable to ask if copper chelation by cuprizone would lead to iron accumulation and iron-mediated loss of oligodendrocytes. Our analysis showed that 60% of oligodendrocytes are lost as early as 2 days after start of the cuprizone diet (Jhelum et al., 2020). Such rapid loss of oligodendrocytes has also been supported by some earlier reports. However, at this timepoint we did not detect increased iron by iron histochemistry or increased ferritin staining. Instead, we found a loss of ferritin staining at day 1, the day before the loss of oligodendrocytes is detected. This decrease in ferritin correlated with increased expression in oligodendrocytes of NCOA4,a cargo receptor that binds to ferritin and shuttles it to autophagosomes for degradation(ferritinophagy) (Mancias et al., 2014; Quiles Del Rey and Mancias, 2019). Interestingly,double immunofluorescence labeling revealed that cells that expressed NCOA4 had low levels or no ferritin, while cells with high levels of ferritin expressed little or no NCOA4.Increased expression of NCOA4 will result in ferritinophagy and the release of bioactive iron, which because of the lack of ferritin will fuel generation of free radicals via the Fenton reaction. Other changes that can add to increase bioactive iron load in oligodendrocytes is reduced expression of the oligodendrocytespecific ferroxidase, hephaestin; reduction of which will lead to reduced iron efflux from oligodendrocytes. In addition, we also detected increased expression of transferrin receptor 1 in oligodendrocytes that will increase iron uptake into these cells. Additionally,intracellular iron load can also be increased by the expression of heme oxygenase-1 that releases iron from heme. The potential rapid increase in iron load from these mechanisms can lead to lipid peroxidation in the CNS (Figure 1). In fact, we detected a rapid increase in lipid peroxidation in the corpus callosum, as seen by a 3-fold increase in 4-hydroxy-2-nonenal, a lipid peroxidation product, as early as 2 days after start of the cuprizone treatment (Jhelum et al.,2020). In addition, double immunofluorescence staining of tissue sections showed that 4-hydroxy-2-nonenal and malondialdehyde,an end-product of polyunsaturated fatty acid peroxidation, are localized to oligodendrocytes in the corpus callosum at this early time period. NCOA4 expression, ferritinophagy, and lipid peroxidation are considered hallmarks of ferroptosis. As mentioned above, another hallmark of ferroptosis is insufficiency of the antioxidant GSH which neutralizes lipid radicals.Two molecules in the GSH pathway were found to be reduced during the first week after start of the cuprizone diet. These include (i)reduction in system Xc, the cystine antiporter,required for influx of cystine into cells for GSH synthesis; and (ii) reduction in GPX4, the enzyme that catalyzes the reaction between GSH and lipid peroxides, and in the process converting reduced GSH to its oxidized from,GSSG. Deficiency in these molecules is also an important feature of ferroptosis (Figure 1).Direct evidence that ferroptosis is responsible for cuprizone-induced loss of oligodendrocytes was obtained from function blocking experiments in which this cell loss was rescued by treatment with a lipid radical scavenging molecule, ferrostatin-1. We also showed that demyelination in the corpus callosum induced by cuprizone can also be rescued by ferrostatin-1. There is a delay of several weeks after the early loss of oligodendrocytes (80%loss by 7 days) before significant myelin loss is seen (4–5 weeks). The slow clearance of myelin,which is also seen in Wallerian degeneration in the CNS, is due to the slow phagocytic response in the adult mammalian CNS. Nevertheless, we show using a novel spectral imaging technique(Nile Red solvatochromism) that there are rapid changes in the molecular composition of myelin with loss of non-polar lipids, as early as 2 days after start of the cuprizone diet(Jhelum et al., 2020). This imaging technique could be very useful to detect early myelin pathology in regions of that look normal with conventional histological techniques, such as in normal appearing white matter in MS, as well as conditions such as stroke, concussion, and other types of CNS trauma.
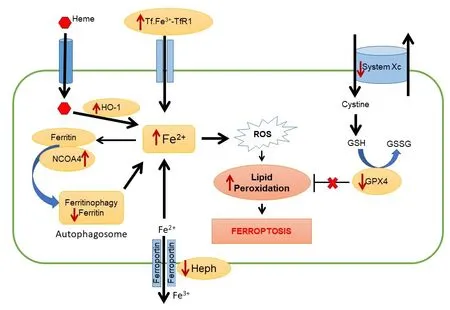
Figure 1|Schematic illustrating key elements of the ferroptosis pathway.
Iron accumulation in the CNS occurs in a variety of neurological conditions and is therefore likely to contribute importantly to neuropathology.To treat these conditions, iron chelators need to be able to cross the blood-brain barrier,enter cells in the CNS to chelate iron, and finally the chelator-iron complex needs to be cleared from the CNS. There is also a need for a better understanding of the mechanisms by which iron mediates cell death, as this could lead to novel alternate therapeutic approaches. The identification and contribution of ferroptosis in various CNS disorders is therefore an important step. It is possible that ferroptosis may occur in some but not all neurological conditions with iron overload. Since iron accumulation is chronic in these disorders, ferroptosis may contribute to only part of the cytotoxic effects and may vary at different stages of the disease from acute to chronic. Ferroptosis may also occur in conditions in which there is no overt iron accumulation (detected by iron histochemistry) but rather a small, transient increase in bioactive iron coupled with reduced iron storage in ferritin, as seen in cuprizone toxicity. The availability of biomarkers that can be used to test cerebrospinal fluid and biopsy samples, as well as autopsy material in neurological disorders will help to make such determinations and pave the way for more effective therapies to treat iron-mediated neurodegeneration.
The present work was supported by a grant from the Canadian Institutes of Health Research(CIHR) (MOP-142231) and MS Society of Canada(MSSOC) (to SD); and MSSOC postdoctoral fellowship (to PJ).
Priya Jhelum, Samuel David*
Centre for Research in Neuroscience and Brain Program, The Research Institute of the McGill University Health Centre, Montreal, QC, Canada
*Correspondence to:Samuel David, PhD,
sam.david@mcgill.ca.https://orcid.org/0000-0002-3314-3695(Samuel David)
Date of submission:December 30, 2020
Date of decision:January 19, 2021
Date of acceptance:March 4, 2021
Date of web publication:June 7, 2021
https://doi.org/10.4103/1673-5374.314300
How to cite this article:Jhelum P, David S (2022)Ferroptosis: copper-iron connection in cuprizoneinduced demyelination. Neural Regen Res 17(1):89-90.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Vikas Mishra, Babasaheb Bhimrao Ambedkar University, India.

- 中国神经再生研究(英文版)的其它文章
- Genes for RNA-binding proteins involved in neuralspecific functions and diseases are downregulated in Rubinstein-Taybi iNeurons
- Research advances on how metformin improves memory impairment in “chemobrain”
- Dendritic spine density changes and homeostatic synaptic scaling: a meta-analysis of animal studies
- Optogenetic activation of intracellular signaling based on light-inducible protein-protein homo-interactions
- Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease
- Growth differentiation factor 5: a neurotrophic factor with neuroprotective potential in Parkinson’s disease