
Lower and upper motor neuron involvement and their impact on disease prognosis in amyotrophic lateral sclerosis
2022-06-13 08:29MariaZakharovaAnnaAbramova
中国神经再生研究(英文版) 2022年1期
Maria N. Zakharova, Anna A. Abramova
Abstract Amyotrophic lateral sclerosis is a fatal neurodegenerative disease characterized by progressive muscle wasting, breathing and swallowing difficulties resulting in patient’s death in two to five years after disease onset. In amyotrophic lateral sclerosis, both upper and lower motor neurons of the corticospinal tracts are involved in the process of neurodegeneration, accounting for great clinical heterogeneity of the disease. Clinical phenotype has great impact on the pattern and rate of amyotrophic lateral sclerosis progression and overall survival prognosis. Creating more homogenous patient groups in order to study the effects of drug agents on specific manifestations of the disease is a challenging issue in amyotrophic lateral sclerosis clinical trials. Since amyotrophic lateral sclerosis has low incidence rates, conduction of multicenter trials requires certain standardized approaches to disease diagnosis and staging. This review focuses on the current approaches in amyotrophic lateral sclerosis classification and staging system based on clinical examination and additional instrumental methods, highlighting the role of upper and lower motor neuron involvement in different phenotypes of the disease.We demonstrate that both clinical and instrumental findings can be useful in evaluating severity of upper motor neuron and lower motor neuron involvement and predicting the following course of the disease. Addressing disease heterogeneity in amyotrophic lateral sclerosis clinical trials could lead to study designs that will assess drug efficacy in specific patient groups, based on the disease pathophysiology and spatiotemporal pattern.Although clinical evaluation can be a sufficient screening method for dividing amyotrophic lateral sclerosis patients into clinical subgroups, we provide proof that instrumental studies could provide valuable insights in the disease pathology.
Key Words: amyotrophic lateral sclerosis; biomarkers of progression; classification;diagnostic biomarkers; disease heterogeneity; electrodiagnostic medicine;electromyography; motor neuron disease; neuroimaging
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal disease characterized by motor neuron degeneration which results in progressive muscle wasting, breathing and swallowing difficulties. The median survival rate varies from two to five years since disease onset (Rinaldi et al., 2017). ALS typically begins focally in one of the limbs, then spreading to the contralateral limb and the adjacent anatomical regions.About 20–30% of all ALS cases have bulbar onset, starting with speaking and swallowing difficulties (Turner et al., 2010;Green et al., 2013). Although skeletal muscles are gradually becoming weaker, until the latest stages of the disease,oculomotor and sphincter muscles remain relatively intact(Sharma et al., 2011). A combination of upper and lower motor neuron signs facilitates the further diagnostic workup.After ruling out any reversible disorders that may mimic ALS,the diagnosis, as well as the further course of the disease,becomes clear (Andersen et al., 2012; Brown and Al-Chalabi,2017).
Nevertheless, in many cases atypical involvement of the upper and motor neurons, various extra-motor manifestations,as well as non-linear course of disease progression, can impede the diagnosis of ALS (Sharma et al., 2011). However,atypical forms of ALS are usually characterized by a more benign course and overall survival prognosis, which makes patients with these ALS variants promising candidates for early therapeutic interventions or recruitment in clinical trials(Jawdat et al., 2015). In this review, we focus on describing the clinical heterogeneity of the disease which stems from different degrees of lower and upper motor neuron involvement.
Search Strategy and Selection Criteria
A search in the National Library of Medicine (PubMed)database was made using the following key words“amyotrophic lateral sclerosis” AND “lower motor neuron” OR“upper motor neuron” AND “pathology”. This search strategy yielded 2678 papers published from 1980 to 2020. For the review, papers concerning the etiopathogenesis, clinical manifestations and disease classification were selected.
Approaches to Amyotrophic Lateral Sclerosis Diagnostics
ALS is a clinical diagnosis that is supported by signs of upper motor neuron (UMN) and lower motor neuron(LMN) involvement during neurological examination and electrodiagnostic testing (nerve conduction studies and needle electromyography (EMG)) in absence of any other clinical abnormalities (de Carvalho et al., 2008; Andersen et al.,2012; Brown and Al-Chalabi, 2017). In case there are certain discrepancies between clinical presentation, instrumental findings and/or past medical history, further neuroimaging or serological studies should be performed, including lumbar puncture and cerebrospinal fluid analysis, in order to exclude any ALS mimics (Andersen et al., 2012).
The majority of ALS patients are being diagnosed with sporadic disease, while only about 5–15% of all cases can be accounted for familial ALS (Mulder et al., 1986; Byrne et al.,2011; Statland et al., 2015).
El Escorial diagnostic criteria (2000) were first suggested as one of the surrogate methods for distinguishing patient groups for clinical trials in ALS. Nevertheless, they soon proved to have very low sensitivity at the earlier stages of the disease,especially in patients with bulbar-onset ALS (Brooks, 2000). In order to solve this issue, additional electrodiagnostic criteria(Awaji-Shima criteria) were introduced in 2006 (de Carvalho et al., 2008). They revised the definition of active denervation:fasciculation potentials in muscles with chronic neurogenic changes on EMG were regarded as signs of active denervation in absence of other spontaneous activity such as fibrillation potentials and positive sharp waves. More importantly,active denervation on EMG was considered an equivalent to clinical signs of LMN involvement. These corrections helped to identify muscles with active denervation that have not yet developed clinically measurable weakness or wasting(Douglass et al., 2010; Krarup, 2011; Amin Lari et al., 2019).Finally, diagnostic category of probable laboratory-supported ALS was omitted (de Carvalho et al., 2008).
Introduction of electrodiagnostic Awaji-Shima criteria led to a substantial improvement in the sensitivity of the diagnostic criteria at the early stages of the disease, without an increase in the number of false-positive results. The proposed Awaji-Shima algorithm showed diagnostic sensitivity of 95% for definite ALS in comparison to the 18% when using clinical El Escorial diagnostic criteria (2000) solely. The increase in diagnostic sensitivity almost doubled in case of bulbar-onset ALS and clinically possible ALS (Carvalho and Swash, 2009;Statland et al., 2015).
Nevertheless, both El Escorial clinical criteria (2000) and Awaji-Shima electrodiagnostic criteria (2006) are mostly used for stratification of ALS patients into subgroups in order justify their inclusion into large multicenter clinical trials (Agosta et al., 2015). In contrast to various functional scales (e.g. ALS Functional Rating Scale – Revised (ALSFRS-R)(Cedarbaum et al., 1999), Appel Scale (Appel et al., 1987)),they do not evaluate the patient’s functional state as well as his need for certain means of palliative care (Hamidou et al.,2017). In order to provide a feasible instrument for rating disease progression with regard to disability, an easy-to-use staging system was proposed, which is based on basic clinical milestones in the natural course of ALS (functional impairment on several anatomical levels and need for gastrostomy and/or respiratory support) (Table 1; Roche et al., 2012).
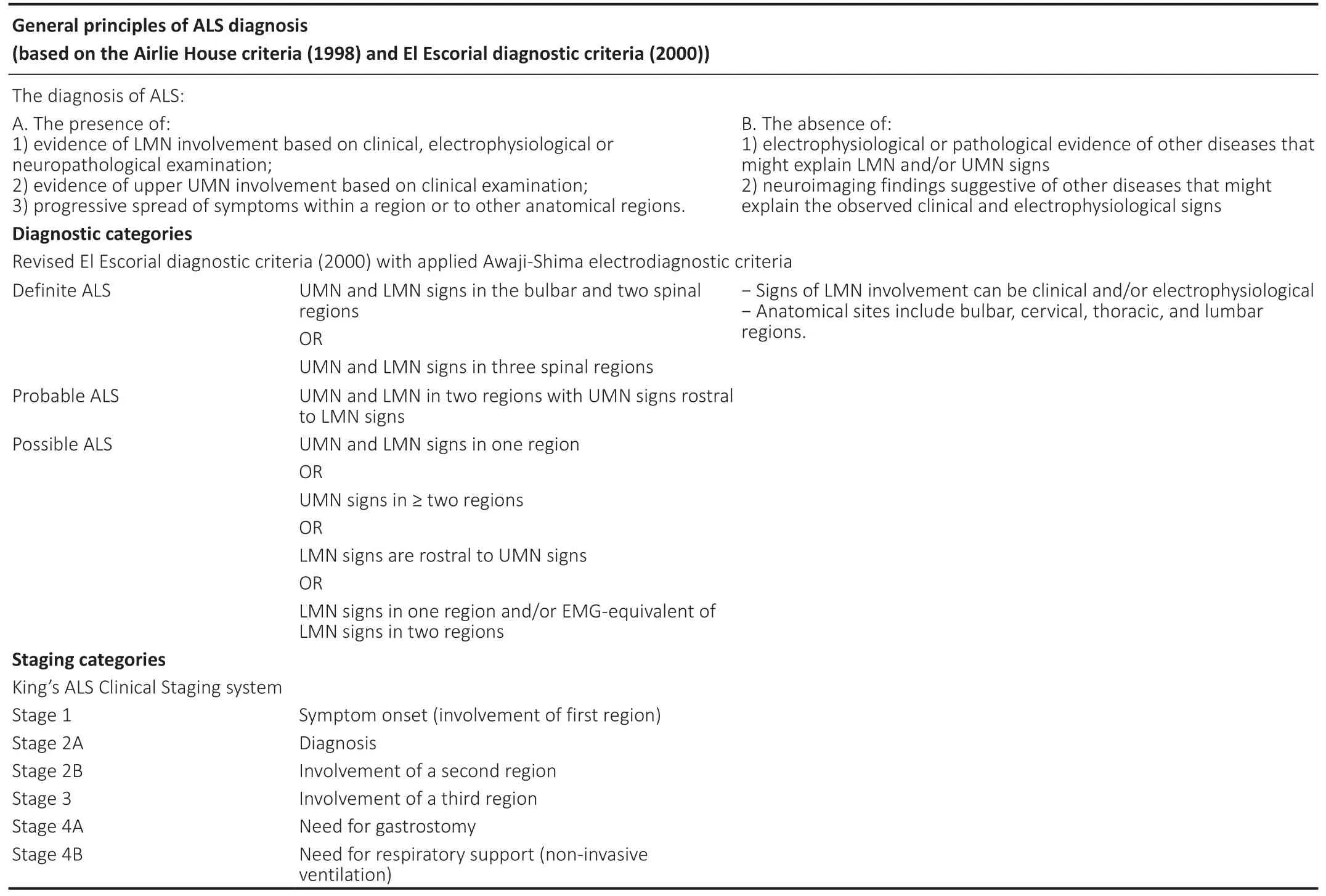
Table 1 |Current approaches to ALS diagnosis and staging
However, several studies have shown that diagnostic categories based on El Escorial diagnostic criteria (2000) can also have prognostic significance (Couratier et al., 2016). For example, in a study by Chio et al. (2002) patients who were diagnosed with definite ALS upon first referral, demonstrated much worse survival rates than those who were diagnosed with probable or possible ALS. In fact, patient groups with probable or possible ALS did not show any statistically significant differences in terms of the following disease progression (Chiò et al., 2002).
Measuring Upper and Lower Motor Neuron Involvement: Diagnostic Capabilities and Prognostic Value
UMN and LMN involvement play an important part in ALS diagnosis and formation of a certain clinical variant of the disease. They also reflect patterns of ALS progression, so their evaluation is essential in order to assess disease progression and evaluate clinical response to certain therapeutic agents if studied longitudinally (Statland et al., 2015; Grad et al., 2017).Neuropathology in ALS includes formation of Bunina bodies and TAR DNA-binding protein 43 (TDP)-positive cytoplasmic inclusions in motor neurons, followed by neuronal loss(Takeda et al., 2020). In addition to astrogliosis and spongiosis,neuroinflammation reactions such as astrocyte activation and proliferation of microglia can be observed both in UMN and LMN. Same reactions are also observed in ALS variants with pure UMN and LMN involvement, confirming pathophysiological unity of various ALS forms (Gordon et al.,2009; Grad et al., 2017).
LMN degeneration is the primary cause for progressive muscle weakness and wasting in ALS, often dominating in the overall clinical presentation of the disease (Dengler,2011; Brown and Al-Chalabi, 2017). Signs of LMN loss can be identified both clinically and instrumentally (Zarei et al.,2015). For diagnostic purposes, only needle EMG is used in order to detect active denervation, which is considered to be a fully-fledged equivalent of LMN involvement in the current diagnostic criteria (de Carvalho et al., 2008). Although many other electrodiagnostic measures and techniques have been suggested so far (Table 2), none of them have yet demonstrated reliable diagnostic capabilities.
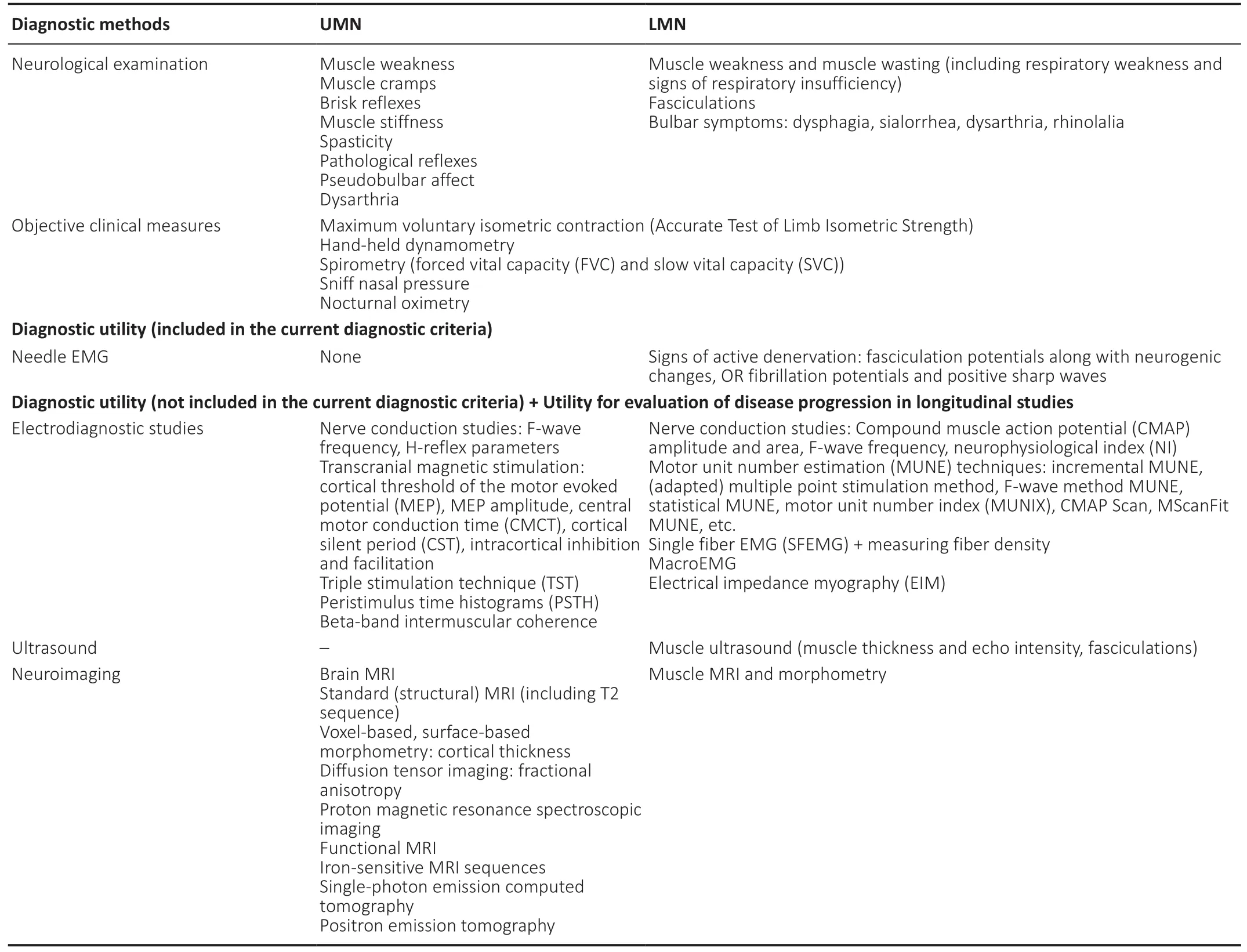
Table 2 |Methods for evaluation of UMN and LMN involvement
Muscle wasting is combined with hyperreflexia, which is usually more pronounced in lower limbs than in upper limbs; in later stages of the disease, brisk tendon reflexes are obscured by widespread and severe muscle atrophy (Zarei et al., 2015; Brown and Al-Chalabi, 2017).
Although many electrophysiological and neuroimaging techniques have demonstrated adequate sensitivity in identifying UMN loss, none of them have been approved as equivalents of clinical signs of UMN involvement (Brooks et al., 2000; Ludolph et al., 2015). Accessible neuroimaging techniques that may point at UMN degeneration include standard brain MRI (T2 sequences, SWI), diffusion tensor imaging, and MRI spectroscopy (Kaufmann and Mitsumoto,2002; Grieve et al., 2016; Huynh et al., 2016; Kassubek and Pagani, 2019). Electrodiagnostic methods that can evaluate the integrity of the corticospinal tracts are single, paired, and triple-pulse TMS (Miscio et al., 1999; Mitsumoto et al., 2007;Floyd et al., 2009; Huynh et al., 2016; Vucic and Rutkove,2018). This is especially important in cases of pure-LMN disease, when clinical UMN signs such as brisk or pathological reflexes can be difficult to elicit (Miscio et al., 1999; Triggs et al., 1999). Some MRI and electrophysiological findings (e.g.hyperintense corticospinal tracts on T2 brain MRI sequence;decreased amplitude of motor evoked potential, prolonged central motor conduction time in TMS) may serve as socalled “supportive” features for the diagnosis of ALS, yet they cannot be fully implemented as specific indicators of UMN degeneration (Miscio et al., 1999; Triggs et al., 1999; Agosta et al., 2015; Huynh et al., 2016).
There are several possible reasons why researchers are reluctant to use instrumental and imaging methods in the current diagnostic criteria as substitutes for clinical UMN signs.Most of these diagnostic techniques have moderate sensitivity and specificity. Specific instrumental findings in patients with motor neuron disease have been reported in limited studies involving small numbers of patients, mostly at late stages of the disease (Agosta et al., 2015; Huynh et al., 2016).Combining multi-modal techniques is a promising solution for obtaining diagnostic work-up algorithms that will have better sensitivity and specificity in terms of identifying UMN loss (Huynh et al., 2016). Only several studies implementing various multi-modal diagnostic methods have been conducted so far. Most of them involved small and heterogenous patient groups, which makes any inter-study comparisons unreliable. Since various neuroimaging and neurophysiological parameters have not shown any straightforward correlation yet, their combinatory use as complementary biomarkers of UMN degeneration has been suggested (Furtula et al., 2013;Grieve et al., 2016; Borsodi et al., 2017).
The pattern of UMN degeneration in ALS is quite different from other known conditions involving neurons of the motor cortex (Swash, 2012). In disorders with UMN lesions (e.g.cerebral infarctions, tumors, multiple sclerosis etc.) not only corticospinal tracts are being influenced by the lesion itself, but the adjacent tracts as well, including rubrospinal,reticulospinal and vestbulospinal projections. This leads to formation of a classical UMN syndrome characterized by pathologically brisk reflexes, repetitive clonus, Babinski sign and spastic muscle tone (velocity-dependent, with a claspknife phenomenon). This type of UMN syndrome is almost never present in ALS, except for pure-UMN variants such as PLS or Mills’ syndrome. In classical Charcot-type ALS, this syndrome is partly obscured not only due to LMN loss, but also because of dysfunction of the supraspinal projections to the α-motor neurons of the anterior horns. Autopsy studies reveal that along with degeneration of α-motor neurons,the number of γ- and β-motor neurons decreases as well(Stephens et al., 2006; Swash, 2012; Álvarez et al., 2018).Thus, the whole segmental motor neuronal structure becomes disrupted, which leads to an ‘alleviated’ UMN syndrome with relatively brisk tendon reflexes, common absence of Babinski sign, or prominent spasticity (Swash, 2012).
Increased cortical excitability in the earlier stages of the disease may account for preliminary mechanisms of compensation in the neurodegenerative process, being part of the typical disease pathophysiology (Andersen et al., 2012;van den Bos et al., 2019).
In a study by Jin et al. (2019), the role of UMN and LMN in functional impairment was studied on three anatomical levels in classical ALS: bulbar, cervical and lumbar. UMN was evaluated based on motor cortical thickness measurement,while spontaneous activity on EMG (fasciculation and fibrillation potentials, positive sharp waves) was regarded as an equivalent for LMN loss. It was demonstrated that bulbar symptoms were derived from an equal degree of LMN and UMN degeneration, while motor dysfunction in upper and lower extremities resulted mostly from LMN loss. Furthermore, patients with bulbar- and cervical-onset ALS exhibited a more pronounced decrease in the motor cortex thickness than lumbar-onset patients, which could be caused by a more prominent functional role of bulbar and arm muscles in the motor homunculus. Epidemiological studies have shown that bulbar-onset classical ALS progresses faster than limb-onset classical ALS, and they both have worse prognosis than progressive muscular atrophy. This fact highlights the possible role of the UMN degeneration in speeding up disease progression in ALS (Chiò et al., 2009;Jawdat et al., 2015; Westeneng et al., 2018).
Current Approaches to Amyotrophic Lateral Sclerosis Classification
Although clinical phenotype in classical ALS (Charcot type) usually consists of obvious signs of UMN and LMN degeneration, current approach to motor neuron diseases classification implies a spectrum of various phenotypes.They can be conditionally divided by the degree of LMN and/or UMN involvement into forms with pure LMN or UMN involvement or a mixture of both (Table 3) (Statland et al.,2015). Among this continuum of motor neuron degeneration,there are forms that are almost restricted to one anatomical region (cervical, lumbar or bulbar), but are also characterized by a specific pattern of LMN and UMN involvement. Thus,there is a considerable overlap between clinical terms which account for different ALS forms (Statland et al., 2015; Al-Chalabi et al., 2016; Grad et al., 2017).
Based on the latest revision of the El Escorial diagnostic criteria (2000) (Ludolph et al., 2015), a diagnosis of ALS can be made if at least the criteria for possible ALS are fulfilled.Thus, restricted phenotypes of ALS include progressive bulbar palsy, brachial amyotrophic diplegia (flail arm, Vulpian-Bernhard, or neurogenic man-in-a-barrel syndrome, also known as scapulohumeral ALS form), flail leg syndrome(Marie-Patrikios’ syndrome, pseudopolyneuritic or peroneal ALS form), progressive muscular atrophy (PMA) and primary lateral sclerosis (PLS) (Grad et al., 2017; Takeda et al., 2020).In all of these disorders, the diagnosis of ALS can be made based on the revised El Escorial diagnostic criteria (2000)(Table 4). Special attention is paid to restricted ALS forms,since all of them develop into generalized ALS with time. With the revised criteria, a diagnosis of possible ALS can be made at earlier stages of these disorders, enabling early recruitment into clinical trials and start of any necessary therapeutic interventions (Ludolph et al., 2015). Nevertheless, it is stressed that restricted forms are characterized by a different prognosis than classical ALS, so their detailed phenotypic classification is also of great importance (Chiò et al., 2011;Talman et al., 2016).
Although PMA and PLS are usually regarded as disorders with pure LMN and UMN involvement respectively, multiple studies have shown that an uneven mixture of LMN and UMN involvement is present both in PMA and PLS (Le Forestier et al., 2001; Statland et al., 2015; Huynh et al., 2016). For example, multiple-point stimulation MUNE in patients with PLS demonstrated lower MUNE values than in control subjects,although they tended to be relatively constant over time(Mitsumoto et al., 2007). These findings are in line with minor denervation and signs of reinnervation which are commonly found in patients with PLS in EMG studies (Le Forestier et al.,2001; Grace et al., 2011; Turner et al., 2020b). Interestingly,those patients with PLS who exhibit very mild signs of active denervation on needle EMG are more likely to transform into classical ALS; in comparison, PLS patients without any signs of active denervation have a much more benign survival prognosis (Gordon et al., 2009; Grad et al., 2017).
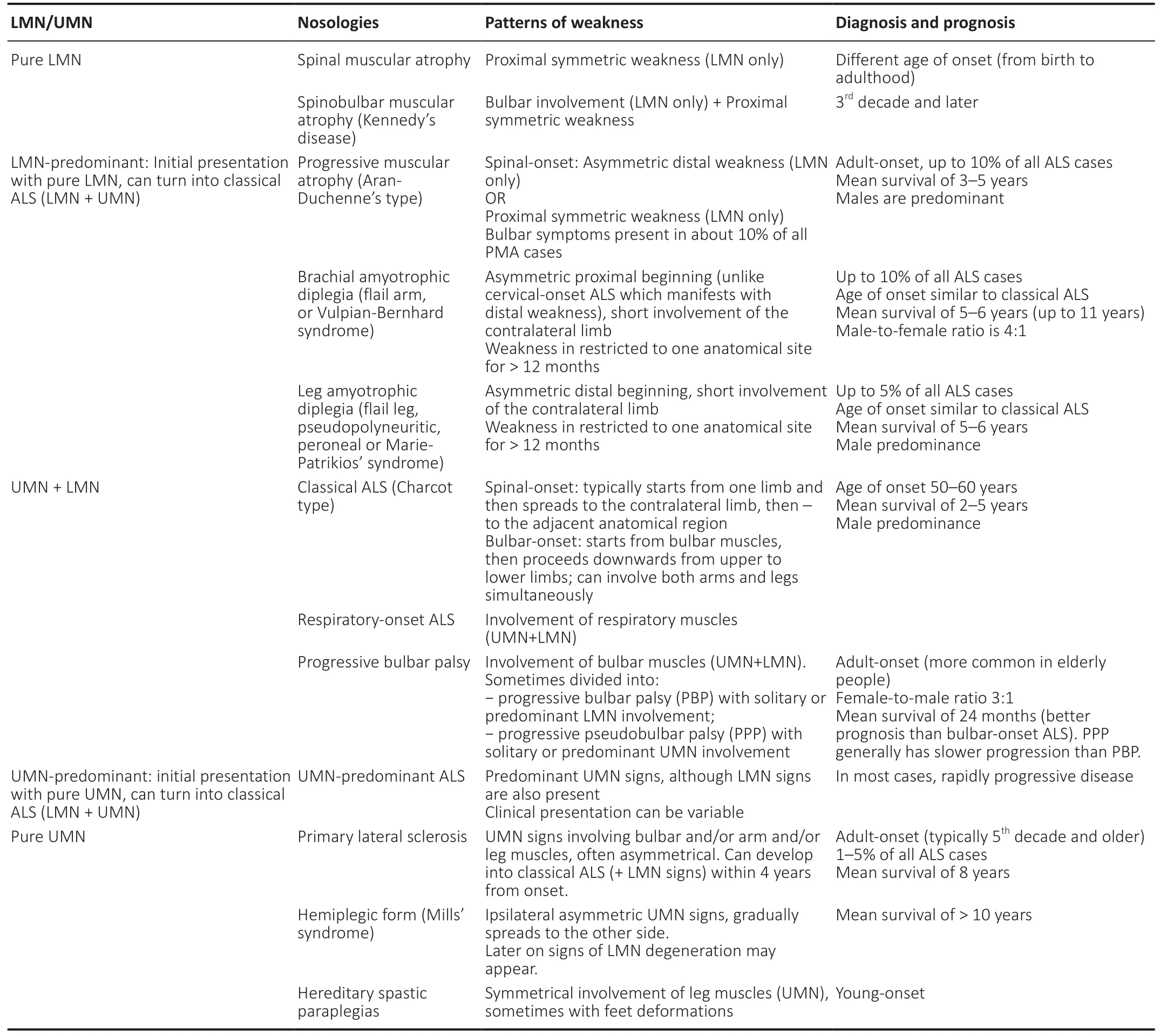
Table 3 |Classification of motor neuron diseases based on the degree of LMN and UMN involvement
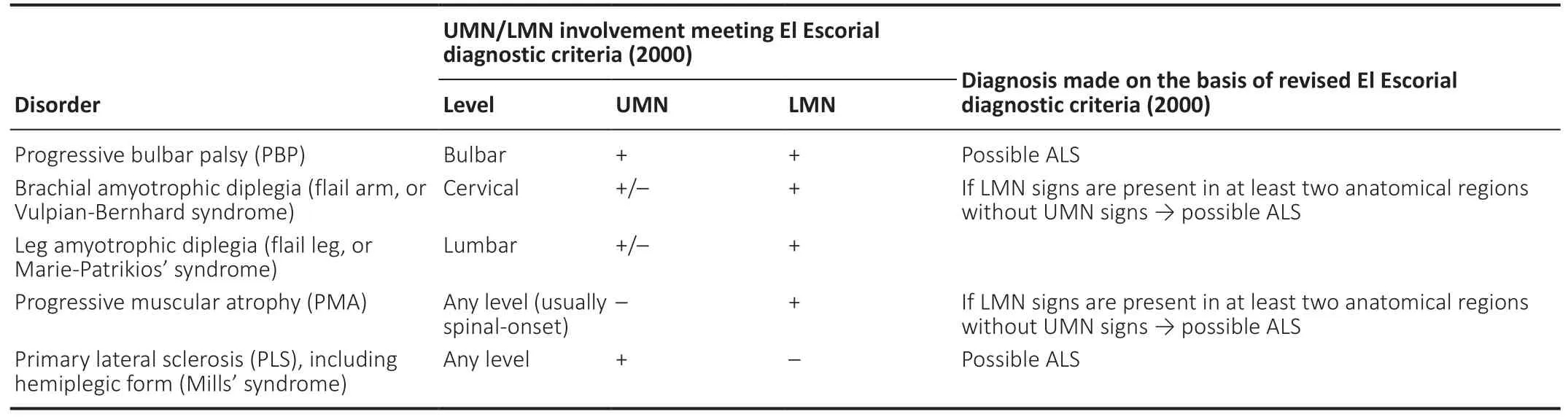
Table 4 |Restricted ALS phenotypes and their relation to El Escorial diagnostic criteria (2000)
In a cohort study by Gordon et al. (2009), more than two thirds (77%) of all patients with pure-UMN ALS developed active denervation by the fourth year after symptom onset.Based on these findings, the following diagnostic criteria for PLS have been proposed. Patients who exhibit UMN signs in at least two out of three anatomical regions (bulbar, cervical and/or lumbar) should be primarily diagnosed with pure-UMN ALS. If progression of UMN involvement in absence of LMN signs persists during the time period of 2–4 years, the patient can be diagnosed with probable PLS. After crossing the time brink of four years, this diagnosis can be changed to definite PLS (Turner et al., 2020a). Nevertheless, cases of deferred transformation of PLS into ALS after 8–27 years since symptom onset have also been described (Bruyn et al., 1995).
On the other hand, multiple postmortem studies have shown UMN degeneration in more than half of all patients with PMA,confirming its direct relationship to the ALS disease spectrum(Ince et al., 2003; Takeda et al., 2020). Evidence of PMA and classical ALS connection can be also found in case reports of familial ALS associated with mutations in superoxide dismutase 1, where same mutations cause both classical ALS and PMA clinical phenotypes in different family members(Appelbaum et al., 1992; Verma and Bradley, 2001).
A distinct PLS variant, which is sometimes considered a separate clinical entity, is hemiplegic ALS, or Mills’ syndrome.It is characterized by progressive unilateral UMN signs in the upper and lower limbs. Several years after disease onset, UMN signs usually spread to contralateral extremities (Turner et al.,2005; Bäumer et al., 2014; Van Laere et al., 2016). This ALS form is very rare, with almost 30 clinical cases described so far, with follow-up ranging from 1.5 to 35 years. Recently, an attempt to propose a diagnostic algorithm for Mills’ syndrome was made, combining clinical, electrophysiological and routine brain neuroimaging data, yet this ALS variant remains a diagnosis per exclusionem. Some cases of Mills’ syndrome later evolve into typical PLS or Charcot-type classical ALS with active denervation (Turner et al., 2005; Jaiser et al., 2019).
In case of progressive bulbar palsy, UMN degeneration(pseudobulbar effect, brisk jaw jerk reflex etc.) is usually slightly predominant in the clinical picture, although it is generally accompanied by tongue atrophy and fasciculations(Karam et al., 2010). This variant is more common in elderly people. It has an overall poorer prognosis in comparison to classical ALS, mostly due to the increased probability of malnutrition and aspiration pneumonia caused by swallowing difficulties (Karam et al., 2010; Zhang et al., 2017; Takeda et al., 2020).
Factors that Influence Disease Phenotype and Prognosis
There are several main factors that determine the clinical phenotype of the disease, including area of onset, extent of lower and upper motor neuron (LMN and UMN, respectively)involvement, spreading pattern, rate of progression on each anatomical level, and age of disease onset (Statland et al., 2015; Couratier et al., 2016). While LMN and UMN degeneration can be detected during routine neurological examination, novel neurophysiological and neuroimaging techniques are useful in their quantitative assessment and detecting pre-clinical motor neuron involvement, shedding light on disease pathophysiology and further prognosis(Neuwirth et al., 2017; Bede and Hardiman, 2018).
Different mutations in ALS-linked genes are also known to be responsible for specific disease phenotypes (including associations with other disorders such as frontotemporal dementia (FTD) and Parkinson disease), age of onset and progression rates. There are many examples of “ALS+”phenotypes which include variable extra-motor symptoms(Verma and Bradley, 2001; Takeda et al., 2020). Although sensory or cerebellar disturbances are supposed to be uncharacteristic for ALS, some of mutations are responsible for ALS with extra-motor manifestations, such as abnormal ocular movement, cerebellar degeneration, sensory impairment, autonomic dysfunction or neurogenic bladder(van der Graaff et al., 2009; McCombe et al., 2017).
Most of ALS patients develop cognitive (up to 75%) and/or behavior disorders (up to 50%) during the disease course.They may vary from executive dysfunction to severe dementia meeting diagnostic criteria for FTD (Huynh et al., 2020).However, neither mild cognitive or behavioral symptoms nor more severe cognitive dysfunction, are included in the El Escorial diagnostic criteria (2000). This drawback is considered to be one of the major weaknesses of the current diagnostic criteria (Agosta et al., 2015; Ludolph et al., 2015; Logroscino,2016).
Unfortunately, data on the possible impact of cognitive and/or behavior dysfunction on disease prognosis are relatively scarce, since these patients are usually omitted from most therapeutic trials. Nevertheless, multiple studies have shown that association with FTD, or moderate to severe cognitive deficit correlate with faster disease progression and shorter survival rates in ALS (Strong et al., 2017; Trojsi et al., 2017;Chiò et al., 2019; Huynh et al., 2020). Behavioral symptoms may lead to low compliance with palliative care, including non-invasive lung ventilation or percutaneous endoscopic gastrostomy, regularity of meal sessions, leading to respiratory insufficiency and progression of malnutrition (Huynh et al.,2020).
Progression rate in ALS highly depends on the disease pathophysiology. In most cases, clinical evaluation reveals focal beginning at a certain anatomical site in the central nervous system, followed by spreading to the adjacent regions and overall generalization. This spreading pattern has been confirmed in multiple studies of focal motor symptoms correlation with localization of the underlying pathology (Ravits et al., 2007; Calvo et al., 2014; Statland et al., 2015). It is undeniable that the clinical course of the disease reflects the pathophysiological processes that occur simultaneously in the CNS (Calvo et al., 2014). That is why the prion-like propagation model remains one of the plausible explanations of the specific disease course in ALS (McAlary et al., 2019). One of the possible mechanisms is formation of “lesions” associated with TDP-43 dysfunction, which later spread gradually along the CNS axis (Gitler and Shorter, 2011;Zufiria et al., 2016). They may occur in the LMN system of the anterior horn of the spinal cord, or in the UMN of the motor cortex, or even in the frontotemporal cortex in case of ALS+FTD phenotypes. For example, the disease can start in the LMN pool, propagating retrogradely towards the UMN pool (“dying-back” hypothesis), or the other way around(Dengler, 2011). Multiple studies aimed at identifying the primary cause of the disease (whether it begins in the motor cortex or anterior hors of the spinal cord) have demonstrated contradictory results, which might be explained by different initiation sites of the disease in CNS. Disease onset may occur in the motor cortex, subcortical structures of the brain, spinal cord, or at multiple sites simultaneously (Vucic et al., 2008;Eisen, 2009; de Carvalho et al., 2011; Dengler, 2011). This theory is supported by uneven incidence of restricted ALS phenotypes in populations of different age and sex, which means that patients who develop certain forms of ALS seem to be susceptible to formation of certain ‘lesions’ of aggregated TDP-43 in the CNS (Takeda et al., 2020). This hypothesis is also supported by better survival prognosis in ALS forms with pure LMN or UMN involvement in comparison to classical ALS.This can be due to relative preservation of the UMN or LMN system, when the most pronounced pathological burden is concentrated on one level of the corticospinal axis (Grad et al., 2017; Takeda et al., 2020).
There are several factors that are generally associated with faster progression rate and worse survival prognosis in ALS(Ludolph et al., 2015; Statland et al., 2015; Huynh et al., 2016;Gaiani et al., 2017; Benatar et al., 2020):
− Pre-morbid state: older age and somatic comorbidities upon disease onset;
− Lower body-mass-index;
− Bulbar-onset ALS;
− Shorter delay to first visit and worse motor function;
− Fast spreading of LMN and UMN symptoms between the first and the second involved anatomical region;− Generalized LMN involvement at the time of diagnosis;− Reduced forced vital capacity at the time of diagnosis;− Moderate to severe cognitive dysfunction (including FTD);− Presence of the C9orf72 repeat expansion;
− Serum and cerebrospinal fluid neurofilament light chains levels.
Regional variants of ALS tend to have a slower rate of progression, since symptoms of LMN and UMN involvement are restricted to a certain anatomical region for a considerable amount of time (Jawdat et al., 2015). Another positive factor in disease progression is predominant involvement of the UMN (Statland et al., 2015).
Current Approaches in ALS Clinical Trials
At the present moment, only two drugs have received FDA approval as therapeutic agents in ALS – riluzole (1995–1996)and edaravone (2017) (Jaiswal, 2019). Both drugs have very moderate influence on disease progression, although their effect may be more profound in case of early therapy initiation and in certain phenotypes of the disease. Although more than sixty therapeutic agents had been tested since riluzole received marketing authorization in Europe in 1996, all of them have shown unsatisfactory clinical efficacy (Petrov et al., 2017).
There are several pitfalls in ALS clinical research which stem from the high clinical and pathogenetic heterogeneity of the disease. For example, genetically engineered ALS animal models cannot represent the full spectrum of various ALS clinical forms in humans (van den Berg et al., 2019). Another issue is that some familial ALS cases can have clinical manifestations and disease course which are similar to sporadic ALS. These patients can be erroneously included in clinical trials for sporadic ALS, while their response to a certain therapeutic agent can be unexpected and lead to inconsistent study results (van den Berg et al., 2019).
Taking into account great clinical, pathophysiological and genetic variability of the disease, as well as absence of any specific diagnostic or prognostic biomarkers, design of clinical trials in ALS should be aimed at including the following enrichment strategies:
(1) Creating more phenotypically homogenous subgroups of patients at earlier stages of the disease that may respond best to a certain therapeutic agent;
(2) Recruiting patients from multiple study sites, which requires standardized approaches to selection process,diagnosis, clinical and instrumental evaluation;
(3) Stratifying patients for possible genetic factors (familial ALS) and signs of behavioral and cognitive impairment that may have a significant influence on disease prognosis;
(4) Incorporating surrogate endpoints, both clinical and instrumental, instead of survival, that will lead to shorter trial length;(5) Implementing emerging neurophysiological, neuroimaging,genetic and biochemical biomarkers in clinical trial design:
(6) Implementing prognostic and prediction models for patients’ stratification (Gladman et al., 2012; Bakkar et al.,2015; Andrews et al., 2019; van den Berg et al., 2019; Goyal et al., 2020).
Conclusion
Addressing disease heterogeneity in ALS clinical trials can lead to study designs that will assess drug efficacy in specific patient subgroups. Every factor that may have an impact on the clinical presentation and further course of the disease should be taken into account, since it provides basis for disease heterogeneity and uneven response to the same treatment interventions. Interference of UMN and LMN signs in the clinical presentation of the disease creates a unique spatiotemporal pattern of the disease. The main goal in ALS research is to gain full comprehension of its pathophysiology based on clinical examination and all accessible instrumental methods, in order to hypothesize which therapeutic agent will be most beneficial for the patient. If we are aiming at acquiring more consistent study results in the short term,including neurophysiological and neuroimaging techniques into upcoming clinical trials in ALS is inevitable. Novel biomarkers might improve existing study designs in ALS clinical trials and provide insight in the pathophysiology of neurodegeneration in ALS.
Author contributions:Supervision, data analysis and interpretation,manuscript drafting: MNZ; data analysis and interpretation, manuscript drafting: AAA. Both authors reviewed and approved the manuscript.
Conflicts of interest:None of the authors have potential conflicts of interest to be disclosed.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Yuri Matteo Falzone, San Raffaele Scientific Institute, Italy; Cinzia Volonté, National Research Council (CNR), Italy.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Genes for RNA-binding proteins involved in neuralspecific functions and diseases are downregulated in Rubinstein-Taybi iNeurons
- Research advances on how metformin improves memory impairment in “chemobrain”
- Dendritic spine density changes and homeostatic synaptic scaling: a meta-analysis of animal studies
- Optogenetic activation of intracellular signaling based on light-inducible protein-protein homo-interactions
- Presenilin mutations and their impact on neuronal differentiation in Alzheimer’s disease
- Growth differentiation factor 5: a neurotrophic factor with neuroprotective potential in Parkinson’s disease