
UPLC-MS/MS法测定食用植物油中乙基麦芽酚含量的不确定度评定
2022-06-07 21:00:06李宣张潆元黄永桥
食品安全导刊 2022年4期
李宣 张潆元 黄永桥






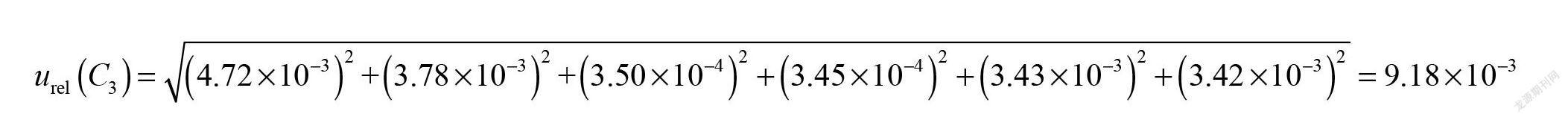


摘 要:依据BJS 201708规定的检测方法,建立数学模型,评定超高效液相色谱-串联质谱法测定食用植物油中乙基麦芽酚含量的不确定度。通过对检测过程中各环节不确定度因素的来源进行分析并对其进行评定,计算合成不确定度。结果表明,测定过程中不确定度的来源主要是标准溶液配制和标准曲线拟合;研究测得植物油中乙基麦芽酚的含量为95.6 μg/kg,其扩展不确定度为4.3 μg/kg(k=2,95%置信区间)。研究可反映测量结果的置信度和准确性,为日常实际检测工作提供技术参考。
关键词:超高效液相色谱-串联质谱法;食用植物油;乙基麦芽酚;不确定度
Uncertainty Evaluation of Determination of Ethyl Maltol in Edible Vegetable Oil by UPLC-MS/MS
LI Xuan1, ZHANG Yingyuan2*,HUANG Yongqiao1
(1.Guizhou Testing Technology Research and Application Center, Guiyang 550014, China; 2.Guizhou Academy of Testing and Analysis, Guiyang 550014, China)
Abstract: According to the detection method specified in BJS 201708, a mathematical model was established to evaluate of the uncertainty of the determination of ethyl maltol in edible vegetable oil by ultra-high performance liquid chromatography tandem mass spectrometry. The sources of uncertainty were analyzed and evaluated, while the components of uncertainty were composed. The sources of uncertainty were comprehensively analyzed and calculated. The results show that the main sources of uncertainty in the determination process are the preparation of standard solution and the fitting of standard curve. The content of ethyl maltol in vegetable oil was 95.6 μg/kg, and the expanded uncertainty is 4.3 μg/kg (k=2, 95% confidence interval). This method accurately reflects the confidence and accuracy of the measurement, which can provide technical references for the daily actual inspection work.
Keywords: ultra-high performance liquid chromatography-tandem mass spectrometry; edible vegetable oil; ethyl maltol; uncertainty
乙基麥芽酚因其具有特殊香味,是一种允许使用的食品用合成香料,作为香味增效剂和香味改良剂被广泛用于烟草、食品、饮料、香料和日用化妆品等行业[1]。乙基麦芽酚具有还原性,可作为抗氧化剂使用,能与金属形成赘合物,可控制人体内铁和铝的含量水平[2-3]。但《食品安全国家标准 食品添加剂使用标准》(GB 2760—2014)附录B中明确规定植物油脂中不得添加食品用香料、香精。一些不法商家为牟取利益,在食用植物油中添加乙基麦芽酚,以次充好。为保障人体健康和安全,需准确检测食用植物油中乙基麦芽酚含量。
目前,乙基麦芽酚的检测方法主要有气相色谱法[4-5]、液相色谱法[6-7]、液相色谱-串联质谱法[8-9]和拉曼光谱法[10]。有关使用液相色谱-串联质谱法测定植物油中乙基麦芽酚的不确定度判定报道不多,测量不确定度是对测量结果质量的定量表征,测量结果的准确性很大程度上取决于其不确定度的大小,建立各种检测的不确定度评定方法,既是提高检测质量的要求,也是实现检测数据国际互认不可或缺的内容[11-12]。研究依据国家市场监督管理总局发布的食品补充检验方法《食用植物油中乙基麦芽酚的测定》(BJS 201708)进行实验,参照现有化学分析中不确定度评定方法和要求,对超高效液相色谱-串联质谱法测定食用植物油中乙基麦芽酚含量的不确定度进行分析评定。根据建立的数学模型,对影响结果的相关分量进行不确定度评估,计算合成相对不确定度,得到最终的扩展不确定度,以期为测量结果的准确性提供科学依据。
1 材料与方法
1.1 材料与试剂
乙腈、甲醇(均为色谱纯,德国Merck公司);甲酸(色谱纯,上海安普实验科技股份有限公司);乙酸铵(优级纯,山东西亚化工科技有限公司);乙基麦芽酚(纯度>99%,上海安谱实验科技股份有限公司)。
1.2 仪器与设备
Agilent 1290超高效液相色谱仪;Agilent 6470 QQQ三重串联四级杆质谱;LT2002电子天平,常熟市天量仪器有限公司;UMV-2多管涡旋混合器,北京普立泰科仪器有限公司;Milli-Q超纯水机,美国Millipore公司;0.22 μm有机滤膜,上海安谱实验科技股份有限公司。
1.3 实验方法
1.3.1 标准溶液配制
①标准储备溶液配制。准确称取乙基麦芽酚标准品101 mg(精确至0.000 1 g),用甲醇溶解并定容至100 mL,得到浓度为1.0 mg/mL标准储备溶液。②标准中间溶液配制。吸取标准储备溶液0.40 mL于100 mL容量瓶中,用甲醇稀释并定容至刻度,得到
4.0 μg/mL标准中间溶液。
1.3.2 仪器条件
(1)液相色谱条件。色谱柱:ZORBAX Eclipse Plus-C18(50 mm×2.1 mm,1.8 μm);流动相A:0.1%甲酸水溶液,流动相B:乙腈;流速:0.3 mL/min;柱温:40 ℃;进样体积:2.0 μL;梯度洗脱程序:0~0.5min,5% B;0.5~2.0 min,5%→90% B;2.0~2.5 min,90% B;2.5~2.8 min,90%→5% B;2.8~3.5 min,5% B。
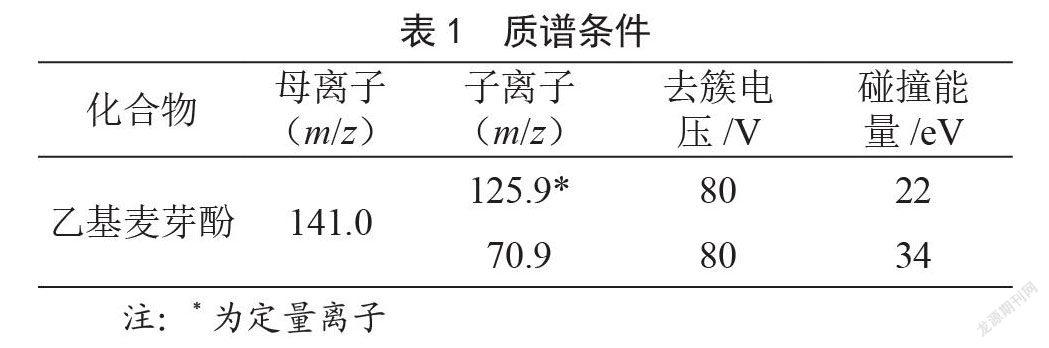
(2)质谱条件。离子源:电喷雾离子源,正离子扫描;多反应监测;毛细管电压:4.5 kV;雾化器压力:40 psi;干燥气流速:10 L/min;干燥气溫度:300 ℃;鞘气流速:10 L/min;鞘气温度:300 ℃。其他质谱条件见表1。
1.3.3 样品前处理
准确称取10 g试样(精确至0.01 g)置于50 mL聚丙烯离心管中,用移液器准确加入10 mL甲醇,涡旋振摇2 min,4 ℃条件下9 000 r/min离心10 min,将上清液移入20 mL具塞刻度试管中,下层油液再用10 mL甲醇重复提取一次,合并上清液,用甲醇定容至20 mL,经微孔滤膜(0.22 μm有机相)过滤,供液相色谱-串联质谱分析。
1.3.4 建立不确定度数学模型
不确定度数学模型如下:
(1)
式中:X为试样中乙基麦芽酚的含量,μg/kg;C为由工作曲线得出的试样溶液中乙基麦芽酚的浓度,ng/mL;V为试样溶液定容体积,mL;m为试样质量,g;为加标回收率修正因子。
2 结果与分析
2.1 不确定度来源
从测量过程和数学模型分析,样品测定的不确定度来源主要有标准溶液的配制、稀释urel(Std)、标准曲线的拟合urel(C)、样品前处理urel(Q)、测量重复性urel(X)和加标回收率urel(R)等。
2.2 标准溶液配制、稀释引入的不确定度urel(Std)
2.2.1 标准品纯度引入的不确定度urel(P)
查标准物质证书,乙基麦芽酚标准物质纯度为99.0%,误差为±0.5%,考虑矩形分布,包含因子k=,属B类评定,则标准品标准不确定度为;标准品相对标准不确定度为。
2.2.2 标准品称量引入的不确定度urel(w)
(1)标准品称量使用万分之一天平,要求称量精确至0.000 1 g。天平检定证书给出的允差为±0.05 mg,考虑矩形分布,包含因子k=,属B类评定,则标准不确定度 mg;相对标准不确定度。
(2)天平重复性标准偏差为±0.02 mg,作为标准品称量重复性不确定度,则相对标准不确定度。
因此,标准物质称量的合成相对标准不确定度为:
2.2.3 标准储备溶液配制引入的不确定度urel(C1)
(1)100 mL单标线容量瓶(A级)引入的不确定度。标准储备液配制使用的100 mL单标线容量瓶(A级),从《常用玻璃量器检定规程》(JJG 196—2006)中可知,100 mL单标线容量瓶(A级)允差为±0.10 mL,考虑三角分布,包含因子k=,属B类评定,则标准不确定度为 mL;相对标准不确定度为。
(2)温度变化引入的不确定度。实验室温度在(20±5)℃变动,甲醇的膨胀系数(20 ℃)为1.18×10-3/℃,对100 mL容量瓶由温度效应产生的体积变化为ΔV =100×5×1.18×10-3 =0.59,
考虑均匀分布,包含因子k=,属B类评定,则标准不确定度为 mL;相对标准不确定度为。
(3)量器使用重复性引入的不确定度。装满溶液至100 mL容量瓶刻度10次,称重并计算标准偏差为0.02 mL,故重复性引入的相对不确定度为。
因此,标准储备溶液配制的合成相对标准不确定度为:
2.2.4 标准中间溶液配制引入的不确定度urel(C2)
(1)1 mL分度吸量管(A级)引入的不确定度。标准中间溶液稀释过程使用1 mL分度吸量管(A级),从《常用玻璃量器检定规程》(JJG 196—2006)中可知,1 mL分度吸量管(A级)容量允差为±0.008 mL,考虑三角分布,包含因子k=,属B类评定,则标准不确定度为 mL;相对标准不确定度为。
(2)温度变化引入的不确定度。实验室温度在(20±5)℃变动,甲醇的膨胀系数(20 ℃)为1.18×10-3/℃,对1 mL分度吸量管(A级)由温度效应产生的体积变化为 ΔV=0.4×5×1.18×10-3=2.36×10-3 mL,考虑均匀分布,包含因子k=,属B类评定,则标准不确定度为 mL;相对标准不确定度为。
(3)100 mL单标线容量瓶(A级)引入的相对标准不确定度为。
(4)100 mL单标线容量瓶(A级)量器使用重复性引入的相对标准不确定度为urel(n2)=2.00×10-4。
(5)100 mL单标线容量瓶(A级)实验室温度变化引起溶剂体积变化引入的相对标准不确定度为。
因此,标准中间溶液配制配制的合成相对标准不确定度为:
2.2.5 标准工作液配制引入的不确定度urel(C3)
实验中,采用阴性试样添加标准溶液,分别吸取0.10 mL、0.20 mL、0.40 mL、0.60 mL、0.80 mL和1.00 mL标准中间溶液加入试样基质中,与试样同时进行提取,制成最终浓度为20.0 ng/mL、40.0 ng/mL、80.0 ng/mL、120.0 ng/mL、160.0 ng/mL和200.0 ng/mL标准系列工作液。配制标准工作液过程移取溶液引入的不确定度来源有使用1 mL分度吸量管(A级)、因实验室温度变化引起的溶剂体积膨胀等。1 mL分度吸量管(A级)按三角分布,包含因子k=;温度变化考虑均匀分布,包含因子k=,配制标准工作液引入的不确定度见表2。因此,阴性试样添加标准溶液1 mL分度吸量管(A级)引入的相对标准不确定度为:
综上,得出标准溶液配制、稀释的合成相对标准不确定度为:
2.3 样品测量重复性引入的不确定度urel(X)
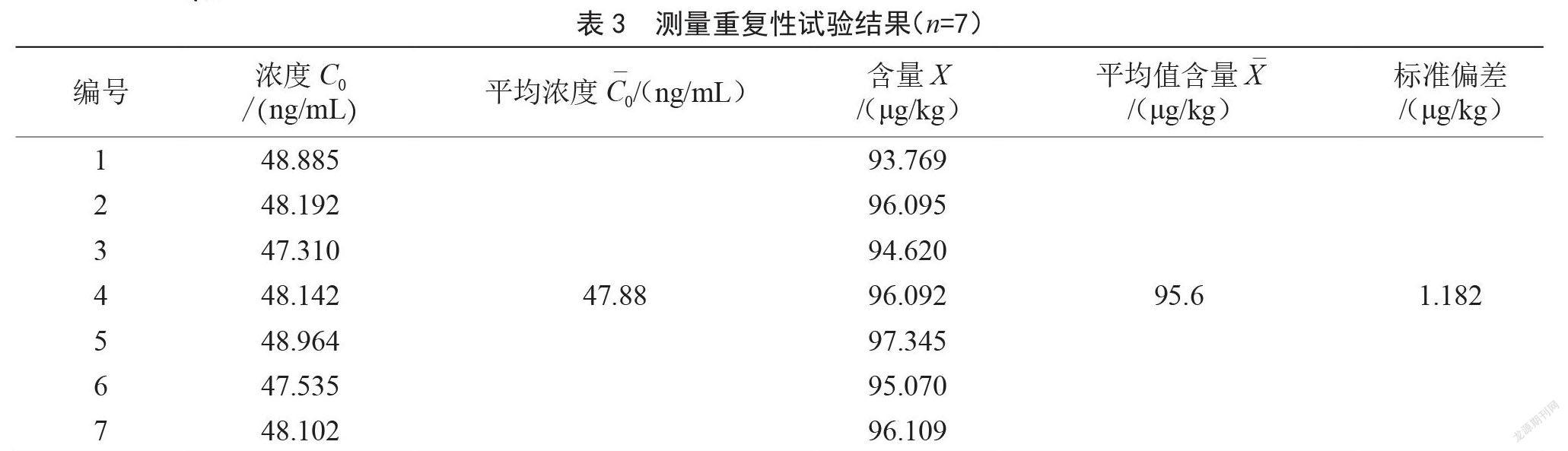
通过7次平行试样检测,计算检测重复性不确定度,此不确定度结果已综合了样品均匀性、前处理、仪器性能等的不确定度,结果见表3,按A类评定计算。重复性标准偏差 μg/kg。重复性标准不确定度 μg/kg。重复性测量相对不确定度。
2.4 标准曲线拟合引入的不确定度urel(C)
使用液相色谱-串联质谱仪对6个不同浓度的标准工作液进行测定,每个质量浓度分别测3次,以质量浓度为横坐标,峰面积为纵坐标,通过直线拟合得到标准曲线,线性回归方程为Y=907.331X-
6 247.866,相关系数r=0.999 7,测定结果見表4,按A类评定计算。
样品重复测定7次,带入线性回归方程计算测定浓度。测得样品中乙基麦芽酚的平均含量为C0=47.88 ng/mL,各标准溶液质量浓度的平均值C=103.33 ng/mL,标准曲线拟合引入的标准不确定度和相对标准不确定度计算公式如下:
(2)
(3)
式中:S为标准溶液峰面积残差的标准差;n为样品平行测定次数,为7;p为标准溶液各浓度点重复测定的次数,为18;C0为所测样品溶液的平均质量浓度,ng/mL;C表示各标准溶液质量浓度点Ci的平均值,ng/mL;k为工作曲线斜率;b为标准曲线截距;Ci为标准溶液各浓度点的含量,ng/mL。
其中,,经计算得到标准曲线拟合引入不确定度u(c)=0.827,相对标准不确定度urel(c)=1.73×10-2。
2.5 样品前处理引入的不确定度urel(Q)
2.5.1 样品称量引入的不确定度urel(m)
称取植物油10 g试样(精确至0.001 g)。天平校准证书给出的允差为±0.005g,考虑矩形分布,包含因子k=,属B类评定,则标准不确定度 g;相对标准不确定度。
2.5.2 样品溶液添加引入的不确定度urel(V)
(1)瓶口分液器引入的不确定度。实验过程中,甲醇提取液的添加使用25.0 mL瓶口分液器。当分液器定值为10.0 mL时其容量允差为±50.0 μL,服从三角分布,包含因子k=,属于B类评定,则标准不确定度 mL;相对标准不确定度。
(2)使用瓶口分液器由温度变化产生的体积变化 ΔV=10×5×1.18×10-3=0.059 mL,考虑均匀分布,包含因子k=,属B类评定,则标准不确定度为 mL;相对标准不
确定度为。瓶口分液器使用2次,因此,样品溶液添加的相对标准不确定度为:
综上,样品前处理引入的合成标准不确定度为:
2.6 回收率引入的不确定度urel(R)
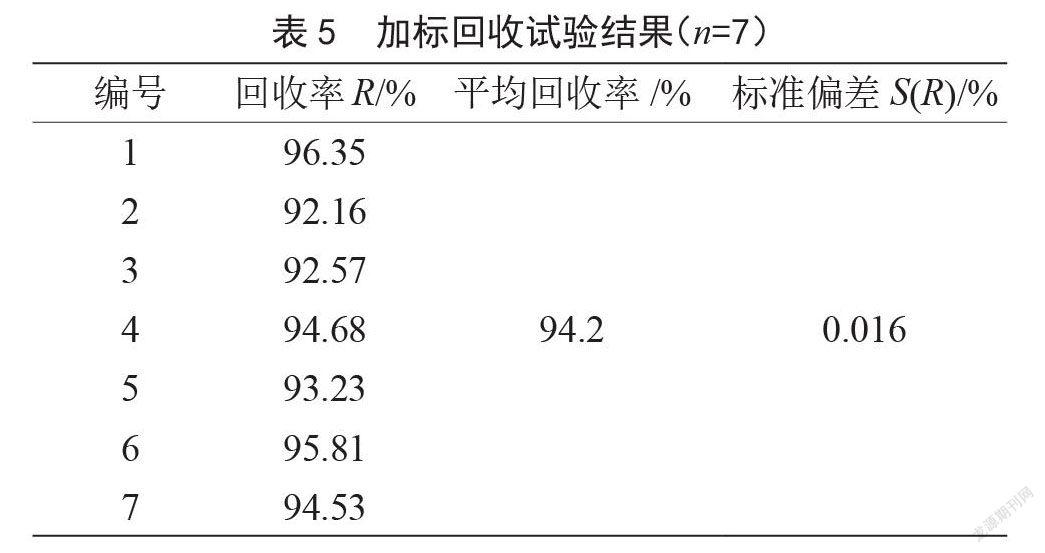
取一阴性样品进行加标回收试验,按1.3.3样品前处理进行处理,平行测定7次,加标回收试验结果见表5。加标回收标准不确定度;加标回收相对不确定度。为确定回收率是否计入乙基麦芽酚含量的计算中,根据《化学分析中不确定的评估指南》(CNAS—GL006:2019),对平均回收率R与1.0进行显著性检验,。查t分布表,取置信区间为95%时,t(0.05,17)>2.447,说明平均回收率R与1.0具有显著差异性,因此在计算公式中必须采用回收率校正因子对结果进行修正。
2.7 合成标准不确定度及扩展不确定度
样品中乙基麦芽酚的测量不确定度各分量见表6。则合成相对标准不确定度为:
依据《化学分析测量不确定度评定》(JJF 1135—2005),按95%置信水平计算测量结果的扩展不确定度,取扩展因子k=2(置信区间为95%),则扩展不确定度为u=k×urel(x)×x=2×0.022 6×95.6=4.3 μg/kg。
2.8 测量结果及不确定度报告
使用超高效液相色谱-串联质谱法测定植物油中乙基麦芽酚含量,根据不确定度判定结果,置信区间为95%时,最终测定结果为X=(95.6±4.3)μg/kg,k=2。
3 结论
采用超高效液相色谱-串联质谱法测定食用植物油中乙基麦芽酚含量,通过对实验过程中引入的不确定度分析量化。结果表明,影响食用植物油中乙基麦芽酚测定结果不确定度的因素主要为标准溶液配制与稀释、标准曲线拟合,样品前处理、测量重复性和加标回收率对不确定度的贡献相对较小。因此,在实际检测过程中要注意操作的规范性、一致性及平行样品的测定问题,以减少测量不确定度,保证检测结果的准确度和可信度。
参考文献
[1]苏国强.乙基麦芽酚生产与应用的新进展[J].食品科学,1988(1):15-20.
[2]穆旻,郑福平,孙宝国,等.麦芽酚和乙基麦芽酚的合成及其在食品工业中的应用[J].中国食品学报,2006,6(1):407-410.
[3]Li Z,LU J L,WU C H,et al.Toxicity Studies of Ethyl Maltol and Iron Complexes in Mice[J].Biomed Research
International,2017,2017:2640619.
[4]景赞,刘超,刘晓碧.气相色谱法测定液态乳中麦芽酚和乙基麦芽酚[J].现代食品,2020(5):199-201.
[5]龚国珍,李媛.气相色谱法测定食用植物油中乙基麦芽酚的含量[J].检验检疫学刊,2019,29(2):136-137.
[6]王浩,刘艳琴,杨红梅,等.高效液相色谱法测定饮料中乙基麦芽酚的研究[J].食品科技,2006(8):235-236.
[7]贺江,马丹露.饮料中乙基麦芽酚UPLC检测方法的建立[J].中国食品添加剂,2018(1):182-186.
[8]马斐,李鑫,张佳慧,等.液相色谱-串联质谱法测定芝麻油中乙基麦芽酚[J].食品安全导刊,2021(26):79-81.
[9]宁霄,何欢,金绍明,等.超高效液相色谱-串联质谱法同时测定食品中麦芽酚、乙基麦芽酚、香兰素、甲基香兰素和乙基香兰素[J].食品安全质量检测学报,2017,8(7):2555-2562.
[10]吴昊,肖东方,应叶,等.基于反应体系的表面增强拉曼光谱快速检测乙基麦芽酚[J].上海师范大学学报(自然科学版),2020,49(2):167-174.
[11]许辉,程刚,周茜,等.禽肉中金刚烷胺残留量测定的不确定度评定[J].食品研究与开发,2013,34(22):68-71.
[12]LEITO S, MOLDER K,KUNNAPAS A,et al.Uncertainty in liquid chromatographic analysis of pharmaceutical product: Influence of various uncertainty sources[J].Journal of Chromatography A,2006,1121(1):55-63.
猜你喜欢
分析化学(2017年1期)2017-02-06 21:34:35
中国纤检(2016年12期)2017-01-20 09:28:19
科技视界(2016年18期)2016-11-03 00:30:59
中国科技博览(2016年22期)2016-11-01 16:48:29
科技视界(2016年21期)2016-10-17 16:41:30
科学与财富(2016年28期)2016-10-14 20:34:01
中国当代医药(2016年2期)2016-03-03 20:29:24
上海医药(2016年1期)2016-02-22 10:32:47
安徽农学通报(2015年10期)2015-06-15 01:20:19
分析化学(2014年3期)2014-12-16 21:38:56
