
烯丙基硫叶立德参与的串联环化反应合成苯并吡喃衍生物
2022-06-01 10:43戴青松杨四琳何美浩刘万聪王亚鹏郭钊宜王启卫1
合成化学 2022年5期
戴青松, 杨四琳, 何美浩, 刘 宇, 刘万聪, 王亚鹏, 郭钊宜, 张 翔*, 王启卫1,,*
(1. 中国科学院 成都有机化学研究所,四川 成都 610041; 2. 成都大学 药学院(川抗所)抗生素研究与再评价四川省重点实验室,四川 成都 610106; 3. 中国科学院大学,北京 100491; 4. 西华大学 理学院,四川 成都 610039)
作为一类重要的含氧杂环化合物,苯并吡喃类衍生物[1]广泛存在于香豆素、黄酮、生物碱等天然产物中,并以其独特的理化性质在医药科学、农业化学、材料化学等领域中体现出重要的应用潜力[2-12](Scheme 1a)。鉴于苯并吡喃衍生物良好的应用价值,开发绿色、简单、高效地构建该类骨架的合成方法[13-19]不仅具有重要的理论意义,也有较好的实际价值。
近年来,衍生自巴豆酸乙酯的烯丙基硫叶立德由于其多样的反应位点,通常在有机合成中作为C1、 C2或C3合成子,参与环化反应用以构建复杂的环状分子骨架[20-36]。围绕其作为C3合成子与α,β-共轭烯酮的反应,研究人员通过底物设计和条件筛选,经[3+3]串联氧杂[2+1][29-30]、 [3+3][31-32]、氧杂[3+4][33-36]等环化过程,分别实现了环己烯并环、苯环、氧杂环庚二烯骨架的构建。然而,通过氧杂[3+3]串联环化反应完成苯并吡喃骨架的构建研究还未见报道,有待进一步探索(Scheme 1b)。
基于本课题组前期对于衍生自巴豆酸乙酯硫叶立德与烯基吲哚亚胺的环化反应[25]及串联反应的研究[37],本文采用酯基烯丙基锍盐作为起始原料,在碱的作用下原位消除形成烯丙基硫叶立德,进而与醌亚胺类化合物发生氧杂[3+3]串联环化反应,以中等到良好的收率合成了一系列苯并吡喃衍生物(Scheme 1c, left)。为进一步简化合成途径、降低合成成本,我们借助“一锅法”的合成策略,以对氨基(羟基)酚类化合物为底物,在碘苯二乙酯(PIDA)的作用下,经其原位氧化串联氧杂[3+3]环化反应,更为高效地实现了目标骨架的构建(Scheme 1c, right)。其代表产物的结构经X-ray单晶衍射进行确证,所得化合物均通过1H NMR,13C NMR和HR-MS(ESI-TOF)进行表征。

Scheme 1
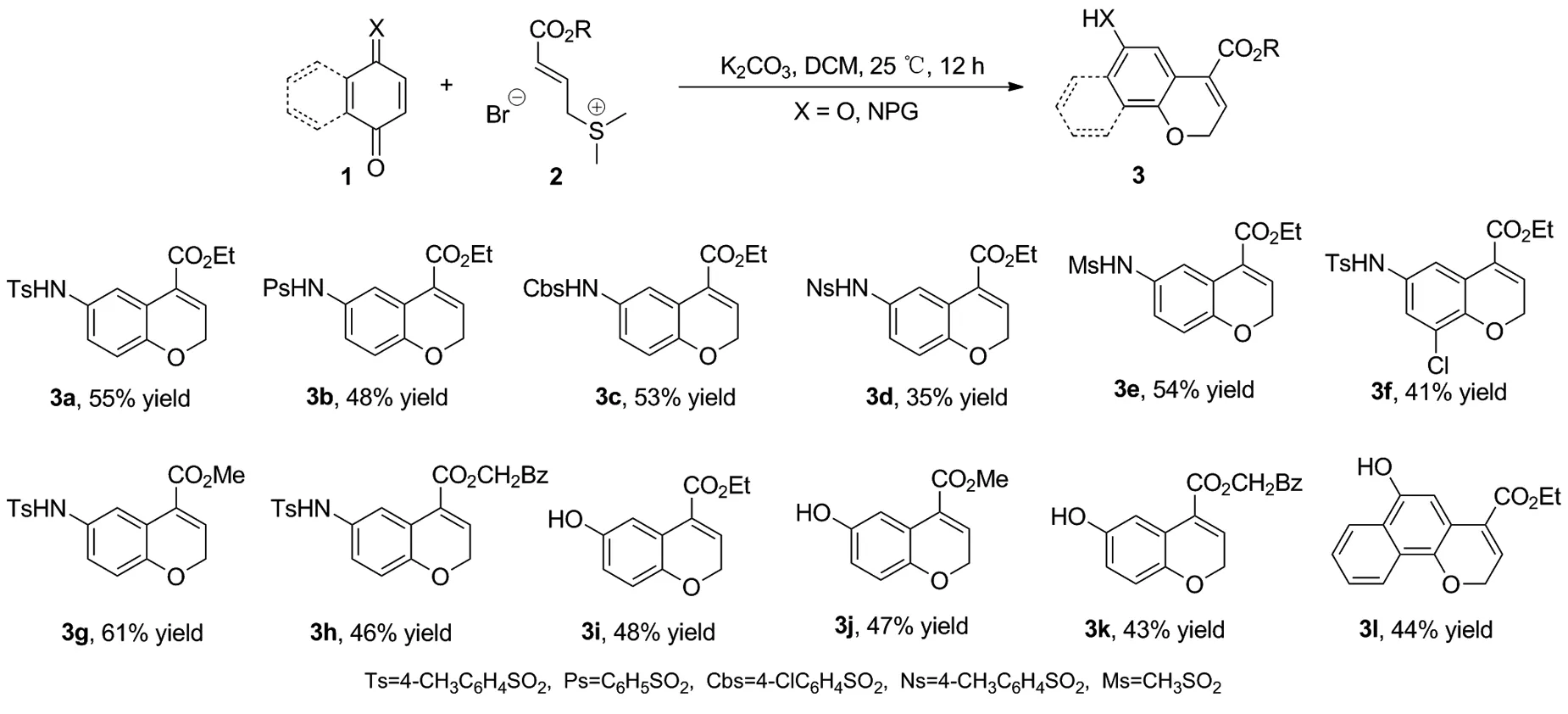
Scheme 2
1 实验部分
1.1 仪器与试剂
WRX-X-4A型熔点仪;JEOL-600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Waters SYNAPT G2型高分辨质谱仪。
醌亚胺1及磺酰基保护的对氨基苯酚4[38]、酯基烯丙基锍盐2[29]按文献方法合成;若无特殊说明,其余所用试剂均为分析纯。
1.2 苯并吡喃衍生物3的合成
(1) 以醌亚胺1为起始原料,合成3的方法A(以3a为例)
在反应试管中依次加入对甲苯磺酰基保护的醌亚胺1a(0.1 mmol, 26.1 mg)、溴巴豆酸乙酯锍盐2a(0.15 mmol, 38.0 mg)及碳酸钾(0.15 mmol, 20.7 mg),加入1.0 mL二氯甲烷,于室温反应12 h(TLC 监测1a消耗完毕)。浓缩,残余物经硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=15/1~5/1]纯化得3a。
用类似的方法合成3b~3l。
(2) 以对氨基(羟基)苯酚4为起始原料,合成3的方法B(以3a为例)
在反应试管中依次加入对甲苯磺酰基保护的对氨基苯酚4a(0.1 mmol, 26.3 mg)、溴巴豆酸乙酯锍盐2a(0.15 mmol, 38.0 mg)、碘苯二乙酯(0.15 mmol, 48.3 mg)及碳酸钾(0.15 mmol, 20.7 mg),加入1.0 mL二氯甲烷,于室温反应12 h(TLC 监测4a消耗完毕)。浓缩,残余物经硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=15/1~5/1]纯化得3a。
用类似的方法合成3b~3l。
6-(4-甲基苯磺酰氨基)-2H-苯并吡喃-4-甲酸乙酯(3a): 淡黄色半固体,方法A收率55%,方法B收率42%;1H NMR(CDCl3, 600 MHz)δ: 7.66~7.61(m, 3H), 7.21(d,J=7.8 Hz, 2H), 6.94(dd,J=8.4 Hz, 2.4 Hz, 1H), 6.82(t,J=3.6 Hz, 1H), 6.71(d,J=8.4 Hz, 1H), 6.62(s, 1H), 4.79(d,J=4.2 Hz, 2H), 4.28(q,J=7.2 Hz, 2H), 2.37(s, 3H), 1.33(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.4, 152.1, 143.6, 136.1, 132.4, 129.9, 129.5, 127.3, 126.8, 124.7, 121.6, 120.0, 116.8, 64.7, 61.1, 21.5, 14.1; HR-MS(ESI-TOF)m/z: calculated for C19H19NO5S{[M+Na]+}396.0876, found 396.0881。
6-(苯磺酰氨基)-2H-苯并吡喃-4-甲酸乙酯(3b): 淡黄色固体,方法A收率48%,方法B收率39%, m.p.129~132 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.75(d,J=7.8 Hz, 2H), 7.62(d,J=3.0 Hz, 1H), 7.52(t,J=7.2 Hz, 1H), 7.46~7.41(m, 2H), 6.94(dd,J=8.4 Hz, 2.4 Hz, 1H), 6.82(t,J=4.2 Hz, 1H), 6.72(d,J=8.4 Hz, 1H), 6.58(s, 1H), 4.80(d,J=4.2 Hz, 2H), 4.27(q,J=7.2 Hz, 2H), 1.33(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.3, 152.3, 139.0, 132.8, 132.5, 129.7, 128.9, 127.3, 126.7, 125.0, 121.8, 120.0, 116.8, 64.8, 61.2, 14.1; HR-MS(ESI-TOF)m/z: calculated for C18H17NO5S{[M+Na]+}382.0720, found 382.0726。
6-(4-氯苯磺酰氨基)-2H-苯并吡喃-4-甲酸乙酯(3c): 淡黄色固体,方法A收率53%,方法B收率40%, m.p.188~192 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.67(d,J=8.4 Hz, 2H), 7.62(d,J=1.8 Hz, 1H), 7.41(d,J=8.4 Hz, 2H), 6.96(dd,J=8.4 Hz, 2.4 Hz, 1H), 6.84(t,J=4.2 Hz, 1H), 6.74(d,J=9.0 Hz, 1H), 6.43(s, 1H), 4.82(d,J=4.2 Hz, 2H), 4.28(q,J=7.2 Hz, 2H), 1.34(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.2, 152.5, 139.4, 137.5, 132.6, 129.3, 129.2, 128.8, 126.5, 125.3, 122.0, 120.1, 117.0, 64.8, 61.2, 14.2; HR-MS(ESI-TOF)m/z: calculated for C18H16ClNO5S{[M+Na]+}416.0330, found 416.0331。
6-(4-硝基苯磺酰氨基)-2H-苯并吡喃-4-甲酸乙酯(3d): 淡黄色固体,方法A收率35%,方法B收率29%, m.p.177~179 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.29(d,J=9.0 Hz, 2H), 7.92(d,J=9.0 Hz, 2H), 7.59(d,J=2.4 Hz, 1H), 7.00(dd,J=9.0 Hz, 2.4 Hz, 1H), 6.85(t,J=4.2 Hz, 1H), 6.76(d,J=9.0 Hz, 1H), 6.51(s, 1H), 4.83(d,J=4.2 Hz, 2H), 4.24(q,J=7.2 Hz, 2H), 1.32(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.0, 153.0, 150.2, 144.7, 132.8, 128.7, 128.4, 126.1, 125.9, 124.2, 122.4, 120.2, 117.2, 64.8, 61.2, 14.1; HR-MS(ESI-TOF)m/z: calculated for C18H16N2O7S{[M+Na]+}427.0570, found 427.0572。
6-(甲磺酰氨基)-2H-苯并吡喃-4-甲酸乙酯(3e): 淡黄色固体,方法A收率54%,方法B收率34%, m.p.97~101 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.86(d,J=2.4 Hz, 1H), 7.15(dd,J=8.4 Hz, 2.4 Hz, 1H), 6.90(t,J=4.2 Hz, 1H), 6.84(d,J=8.4 Hz, 1H), 6.61(s, 1H), 4.84(d,J=4.2 Hz, 2H), 4.31(q,J=7.2 Hz, 2H), 2.98(s, 3H), 1.36(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.4, 152.4, 132.9, 129.9, 126.5, 124.8, 121.3, 120.3, 117.2, 64.8, 61.2, 39.0, 14.2; HR-MS(ESI-TOF)m/z: calculated for C13H15NO5S{[M+Na]+}320.0563, found 320.0565。
8-氯-6-(4-甲基苯磺酰氨基)-2H-苯并吡喃-4-甲酸乙酯(3f): 淡黄色固体,方法A收率41%,方法B收率32%, m.p.142~145 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.66(d,J=8.4 Hz, 2H), 7.59(d,J=3.0 Hz, 1H), 7.24(d,J=8.4 Hz, 2H), 7.08(d,J=3.0 Hz, 1H), 6.88(t,J=3.0 Hz, 1H), 6.64(s, 1H), 4.91(d,J=3.6 Hz, 2H), 4.28(q,J=7.2 Hz, 2H), 2.39(s, 3H), 1.34(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.0, 147.7, 144.0, 135.8, 133.0, 130.0, 129.7, 127.3, 126.4, 124.4, 121.6, 121.1, 119.4, 65.3, 61.4, 21.5, 14.1; HR-MS(ESI-TOF)m/z: calculated for C19H18ClNO5S{[M+Na]+}430.0486, found 430.0488。
6-(4-甲基苯磺酰氨基)-2H-苯并吡喃-4-甲酸甲酯(3g): 淡黄色固体,方法A收率61%,方法B收率45%, m.p.148~152 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.63(d,J=8.4 Hz, 2H), 7.60(d,J=3.0 Hz, 1H), 7.22(d,J=7.8 Hz, 2H), 6.95(dd,J=8.4 Hz, 3.0 Hz, 1H), 6.83(t,J=4.2 Hz, 1H), 6.71(d,J=8.4 Hz, 1H), 6.63(s, 1H), 4.79(d,J=4.2 Hz, 2H), 3.81(s, 3H), 2.37(s, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.8, 152.1, 143.6, 136.1, 132.8, 129.9, 129.6, 127.4, 126.5, 124.7, 121.6, 119.9, 116.8, 64.7, 52.1, 21.5; HR-MS(ESI-TOF)m/z: calculated for C18H17NO5S{[M+Na]+}382.0720, found 382.0716。
6-(4-甲基苯磺酰氨基)-2H-苯并吡喃-4-甲酸(2-氧代-2-苯乙基)酯(3h): 淡黄色固体,方法A收率46%,方法B收率37%, m.p.129~132 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.94(dd,J=8.4 Hz, 1.2 Hz, 2H), 7.67~7.60(m, 4H), 7.55~7.50(m, 2H), 7.18(d,J=7.2 Hz, 2H), 7.02~6.99(m, 2H), 6.73(d,J=9.0 Hz, 1H), 6.55(s, 1H), 5.48(s, 2H), 4.83(d,J=4.8 Hz, 2H), 2.33(s, 3H);13C NMR(CDCl3, 150 MHz)δ: 191.8, 163.7, 152.0, 143.6, 136.0, 134.1, 134.0, 133.8, 130.0, 129.5, 129.0, 127.8, 127.3, 125.8, 124.8, 121.5, 119.7, 116.8, 115.9, 66.3, 64.8; HR-MS(ESI-TOF)m/z: calculated for C25H21NO6S{[M+Na]+}486.0982, found 486.0985。
6-羟基-2H-苯并吡喃-4-甲酸乙酯(3i): 淡黄色半固体,方法A收率48%,方法B收率35%;1H NMR(CDCl3, 600 MHz)δ: 7.50(d,J=3.0 Hz, 1H), 6.88(t,J=4.2 Hz, 1H), 6.75(d,J=8.4 Hz, 1H), 6.69(dd,J=8.4 Hz, 3.0 Hz, 1H), 4.81(s, 1H), 4.76(d,J=4.2 Hz, 2H), 4.31(q,J=7.2 Hz, 2H), 1.36(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 164.8, 150.0, 147.9, 132.7, 127.1, 120.4, 116.9, 116.3, 113.0, 64.6, 61.1, 14.2; HR-MS(ESI-TOF)m/z: calculated for C12H12O4{[M+Na]+}243.0628, found 243.0631。
6-羟基-2H-苯并吡喃-4-甲酸甲酯(3j): 淡黄色半固体,方法A收率47%,方法B收率40%;1H NMR(CDCl3, 600 MHz)δ: 7.49(d,J=2.4 Hz, 1H), 6.88(t,J=4.2 Hz, 1H), 6.76(d,J=9.0 Hz, 1H), 6.69(dd,J=8.4 Hz, 2.4 Hz, 1H), 5.00(s, 1H), 4.76(d,J=4.2 Hz, 2H), 3.85(s, 3H);13C NMR(CDCl3, 150 MHz)δ: 165.2, 150.0, 147.9, 133.1, 126.9, 120.2, 117.0, 116.4, 113.0, 64.6, 52.1; HR-MS(ESI-TOF)m/z: calculated for C11H10O4{[M+Na]+}229.0471, found 229.0479。
6-羟基-2H-苯并吡喃-4-甲酸(2-氧代-2-苯乙基)酯(3k): 淡黄色固体,方法A收率43%,方法B收率25%, m.p.116~121 ℃;1H NMR(CDCl3, 600 MHz)δ: 7.96(d,J=7.2 Hz, 2H), 7.64(t,J=7.2 Hz, 1H), 7.52(d,J=7.8 Hz, 2H), 7.04(t,J=4.2 Hz, 1H), 6.76(d,J=9.0 Hz, 1H), 6.70(dd,J=8.4 Hz, 2.4 Hz, 2H), 5.52(s, 2H), 5.11(s, 1H), 4.79(d,J=4.2 Hz, 2H);13C NMR(CDCl3, 150 MHz)δ: 192.2, 164.2, 150.1, 147.8, 134.1, 134.0, 129.0, 127.8, 126.7, 120.0, 117.0, 116.5, 116.2, 113.1, 66.3, 64.6; HR-MS(ESI-TOF)m/z: calculated for C18H14O5{[M+Na]+}333.0733, found 333.0726。
6-羟基-2H-苯并色烯-4-甲酸乙酯(3l): 淡黄色半固体,方法A收率44%,方法B收率29%;1H NMR(CDCl3, 600 MHz)δ: 8.14~8.08(m, 2H), 7.54(s, 1H), 7.48~7.45(m, 2H), 6.88(t,J=4.2 Hz, 1H), 6.02(s, 1H), 4.91(d,J=4.2 Hz, 2H), 4.33(q,J=7.2 Hz, 2H), 1.37(t,J=7.2 Hz, 3H);13C NMR(CDCl3, 150 MHz)δ: 165.2, 145.6, 144.0, 130.7, 127.8, 126.1, 126.1, 125.2, 125.1, 122.0, 121.8, 114.4, 105.6, 64.8, 61.2, 14.2; HR-MS(ESI-TOF)m/z: calculated for C16H14O4{[M+Na]+}293.0784, found 293.0788。
2 结果与讨论
2.1 醌亚胺参与的氧杂[3+3]串联环化反应的条件筛选
以3a的合成反应为模板反应,对反应条件进行了优化(表1)。首先以碳酸铯为碱,在室温下对反应溶剂进行了筛选(Entry 1~4)。结果发现,相比于其他溶剂,当以二氯甲烷为反应溶剂时,反应能够较好的进行(Entry 3)。基于这一结果,进一步在二氯甲烷作溶剂的条件下,对反应所需的碱进行了优化(Entry 5~10)。结果显示,碱性较强(Entry 8)或较弱(Entry 5)都不利于反应的发生;当使用亲核性较强的DABCO时,反应较为杂乱,几乎无法分离得到目标产物(Entry 9);最终确定了碱性适中的碳酸钾作为反应的最优碱。最后,探索了温度对于反应的影响(Entry 11~12)。结果表明,当降低温度时,反应效率出现了较大程度的下降(Entry 11);而升高反应温度至50 ℃时,TLC显示反应较为杂乱(Entry 12)。因此,确定了反应的最优条件:使用二氯甲烷作溶剂,以碳酸钾作为碱,于室温下进行反应。

表1 氧杂[3+3]串联环化反应的优化Table 1 Optimization of the oxa-[3+3] annulation
2.2 醌亚胺参与的氧杂[3+3]串联环化反应的底物扩展
在优化的条件下,对反应的底物普适性进行了研究(Scheme 2)。首先以含有不同取代基的醌亚胺为底物进行反应,均能以中等的收率分离得到目标化合物3a~3f,最高可达55%(3a)。随后的实验结果表明,反应对于烯丙基硫叶立德上的取代基也同样具有较好的耐受性(3g~3h)。接下来,我们进一步对醌及萘醌的底物兼容性进行了考查,发现反应同样能够顺利的发生(43%~48%),得到相应的目标产物(3i~3l)。值得一提的是,当以萘醌作为底物时,这一氧杂[3+3]串联环化反应为氧杂菲衍生物的高效合成提供了一种绿色、高效的合成方法。
2.3 对氨基(羟基)苯酚参与的氧化串联氧杂[3+3]环化反应的条件筛选
为进一步简化合成途径、降低合成成本,进一步探索了以对甲苯磺酰基保护的氨基苯酚为底物,经原位氧化串联氧杂[3+3]环化反应的“一锅法”合成策略。通过对反应溶剂(Entry 1~4),碱(Entry 5~10)以及氧化剂(Entry 11~13)的筛选,我们确定了该氧化串联氧杂[3+3]环化反应的最优条件:使用二氯甲烷作为溶剂,以碳酸钾作为碱,利用碘苯二乙酯对酚类化合物进行原位氧化,进而发生氧杂[3+3]环化反应。

表2 3a“一锅法”合成反应条件的优化Table 2 Optimization of the “one-pot” synthesis condition of 3a
2.4 对氨基(羟基)酚参与的氧化串联氧杂[3+3]环化反应的底物扩展
在“一锅法”合成的优化条件下,对反应的底物普适性进行了研究(表3)。实验结果表明,无论是以含有不同取代基的对氨基苯酚(Entry 1~6),还是以不同酯基取代的烯丙基锍盐为底物进行反应时(Entry 7~8),尽管收率相较于以醌亚胺为底物时均出现了不同程度的下降,但反应均能顺利的发生,得到相应的目标产物(3a~3h)。随后,苯酚及萘酚底物兼容性的考查结果也进一步论证了该方法的普适性(Entry 9~12)。

表3 “一锅法”合成反应的底物扩展Table 3 Substrate scope of the “one-pot” synthesis
2.5 苯并吡喃衍生物的结构确证
通过对产物3f进行X射线单晶衍射实验,确证了产物的分子结构(图1)。其余化合物的分子结构可通过类推得到。

图1 化合物3f的单晶结构(CCDC: 2092870)Figure 1 The crystal structure of compound 3f
2.6 反应机理
根据实验结果与文献报道[39],提出了该反应可能的反应机理(Scheme 3)。首先,对甲苯磺酰氨基苯酚4a在碘苯二乙酯的作用下,被氧化成对甲苯磺酰基保护的醌亚胺1a。同时,巴豆酸乙酯衍生的锍盐在碱的作用下消除得到烯丙基硫叶立德I,经电荷转移后得到中间体II。随后,中间体II进攻1a,经质子转移(PT)得到两性离子中间体III。接下来,中间体III借助芳构化的动力完成烯醇互变得到酚氧负离子中间体IV,并再次通过质子转移(PT)得到两性离子中间体V。最后,中间体V通过分子内SN2亲核取代反应脱去二甲基硫醚并得到目标环化产物3a。

Scheme 3
在室温下,以二氯甲烷作为溶剂,采用酯基烯丙基锍盐作为起始原料,在碳酸钾的作用下原位消除形成烯丙基硫叶立德,与醌亚胺类化合物经氧杂[3+3]串联环化反应,以中等的收率合成了一系列苯并吡喃衍生物。随后,借助“一锅法”的合成策略,以对氨基(羟基)酚类化合物为底物,在碘苯二乙酯(PIDA)的作用下,经原位氧化串联氧杂[3+3]环化反应同样实现了目标骨架的构建,进一步简化了合成途径、降低了合成成本。为后续此类苯并吡喃骨架的高效构建提供了两种绿色、高效的合成方法,相关深度研究仍在进行中。
猜你喜欢
分析测试学报(2022年10期)2022-10-22
酿酒科技(2022年8期)2022-08-20
核化学与放射化学(2022年2期)2022-04-28
检验医学(2022年2期)2022-03-14
合成化学(2022年1期)2022-02-19
饲料博览(2020年7期)2020-08-18
药学研究(2015年11期)2015-12-19
安徽化工(2015年4期)2015-12-12
分析化学(2014年7期)2014-12-13
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
